线粒体分裂在肺动脉高压肺血管重构中的作用*
2022-05-06蒋智渊林葆菁黄荣杰
蒋智渊, 林葆菁, 黄荣杰
(广西医科大学第一附属医院心血管内科,广西 南宁 530021)
肺动脉高压(pulmonary hypertension,PH)是一类以肺血管阻力进行性升高为主要特征的疾病,其发病率约为每年2.0~7.6/100 万,患病率则为11~26/100万[1]。PH致残、致死率高,预后差,是目前心血管疾病治疗的难点之一[1]。目前治疗PH 的药物包括L型钙离子通道拮抗剂、磷酸二酯酶抑制剂、可溶性鸟苷酸环化酶激动剂、内皮素受体拮抗剂和前列环素激动剂。上述药物主要针对肺血管收缩,而对于PH 发病中最重要的环节——肺血管重构的作用有限[1-2]。血管内皮细胞功能紊乱和平滑肌细胞增生是肺血管重构的中心环节[2]。肺血管重构发生的机制复杂,涉及骨形态发生蛋白受体2 信号通路受损[3]、生长因子信号通路异常激活(如血管内皮生长因子信号通路和血小板源生长因子信号通路)[4-5]、离子通道功能异常(如钾通道亚家族 K 成员 3)[6]、炎症损伤[7]、氧化应激[7]、能量代谢异常[8]等。线粒体是机体最重要的产能场所,线粒体功能异常不仅引起肺动脉内皮细胞(pulmonary artery endothelial cells,PAECs)和肺动脉平滑肌细胞(pulmonary artery smooth muscle cells,PASMCs)能量代谢障碍,还可以产生大量活性氧簇(reactive oxygen species,ROS),增加氧化应激和激活炎症反应,因此线粒体功能障碍与PH 的发生密切相关[9]。线粒体分裂是线粒体动力代谢的重要组成部分,它与线粒体融合一起,动态维持着线粒体正常功能。本文将从线粒体分裂在肺血管重构中的作用进行阐述,以期为PH的治疗提供新的思路。
1 线粒体动力代谢的过程
线粒体是细胞内最重要的供能场所,线粒体通过不断融合与分裂维持代谢平衡。轻度能量缺乏时,线粒体融合成管状网络使产能最大化,满足机体需要。而当持续、严重的应激时,线粒体发生分裂,产生大量线粒体碎片和ROS,对蛋白、脂质和线粒体DNA造成损害,造成能量供应障碍[10]。
线粒体分裂时首先通过与内质网接触,发生收缩反应,接着发动蛋白相关蛋白1(dynamin-related protein 1,DRP1)在DRP1 适配体——线粒体分裂因子(mitochondrial fission factor,MFF)、线粒体动力蛋白49 kD(mitochondrial dynamics protein of 49 kD,MiD49)和MiD51的协同下,被招募到线粒体外膜,促进线粒体外膜的收缩和分裂,从而启动线粒体分裂的过程。线粒体融合则由相邻线粒体通过外膜和内膜的融合完成。线粒体融合蛋白1(mitofusin 1,MFN1)和MFN2 介导线粒体外膜的融合,视神经萎缩蛋白1(optic atrophy 1,OPA1)则介导线粒体内膜的融合[11]。
2 线粒体分裂与PH
2.1 线粒体分裂与PASMCs PASMCs 增殖和凋亡抵抗导致肺动脉收缩和狭窄,是PH 发生的重要原因。Marsboom 等[12]发现 PH 患者 PASMCs 线粒体分裂明显增加,且细胞增殖明显,通过抑制线粒体分裂可有效的减缓 PH 患者 PASMCs 增殖。Zhang 等[13]发现缺氧诱导的PH 大鼠模型中,肺动脉中层肥厚,且缺氧诱导的大鼠PASMCs 线粒体分裂明显增加。Parra 等[14]也发现低氧诱导人 PASMCs 线粒体分裂,促进PASMCs 增殖,抑制线粒体分裂可以减少PASMCs增殖。在注射野百合碱构建的PH大鼠模型中,同样存在着线粒体过度分裂所致的PASMCs 增殖现象[15]。此外,Wang 等[16]发现慢性肺血栓性 PH患者PASMCs 线粒体过度分裂,出现去分化和明显的增殖,抑制线粒体分裂可以阻止上述情况的发生。上述研究提示,线粒体过度分裂所致的PASMCs 增殖在PH的发生中起着重要作用。
2.2 线粒体分裂与PAECs PAECs 功能异常、迁移能力增加、异常增殖和凋亡抵抗是肺血管梗阻性病变形成的重要原因,与肺动脉压力增高密切相关[17-18]。PH 患者PAECs 增殖和迁移能力明显增强,提示PAECs 增殖和迁移异常在PH 的发生中起着重要作用[17]。Suresh 等[19]利用 SU5416/低氧构建 PH 大鼠模型,发现PH大鼠PAECs增殖和迁移能力增强,并且伴有线粒体过度分裂。Shen等[20]发现,缺氧诱导PAECs线粒体分裂增加,细胞增殖和迁移能力明显增强,抑制线粒体分裂,可减弱PAECs增殖和迁移。而在缺氧诱导的小鼠模型中,线粒体分裂增加,与肺新生血管增多有关,抑制线粒体分裂可以减少肺血管新生,减轻右心室收缩压和右室肥厚。上述研究表明,线粒体过度分裂促进PAECs的增殖与迁移,导致PH的发生。
3 PH中线粒体分裂的调控靶点
3.1 DRP1 DRP1 在线粒体分裂的调控中起着至关重要的作用,在各种病理因素刺激下,DRP1 由细胞浆转移至线粒体外膜,促进线粒体外膜的收缩和分裂,从而启动线粒体分裂的过程。已有大量的证据表明,下调DRP1 可以逆转PH 的形成。Marsboom等[12]发现 PH 患者 PASMCs 中 DRP1 表达明显升高,第616 位点丝氨酸磷酸化比例增加,线粒体的分裂明显增加,同时细胞的增殖显著增加。采用DRP1抑制剂Mdivi-1 或RNA 干扰下调DRP1 表达,可以显著减少PH 患者PASMCs 的增殖。他们同时利用慢性缺氧、二氯化钴或野百合碱腹腔内注射构建大鼠PH模型。他们发现,上述PH 模型中PASMCs 线粒体分裂增加,均与缺氧诱导因子1(hypoxia-inducible factor-1,HIF-1)激活,通过 cyclin B1/CDK1 信号通路,上调DRP1 的表达和DRP1 第616 位点丝氨酸磷酸化比例有关,通过Mdivi-1 抑制DRP1 活性,可以减少上述动物模型PASMCs 增殖,改善活动能力和右心室功能。Zhang 等[13]发现缺氧诱导的 PH 大鼠模型中,ROS 和线粒体来源的ROS,通过上调DRP1,促进线粒体分裂,从而导致PASMCs 增殖,PASMCs 凋亡减少。而通过下调DRP1表达,则可以减少缺氧诱导的PASMCs 增殖。Parra 等[14]的研究则提示,可以通过抑制DRP1 活性或负性突变抑制DRP1 的功能,减少线粒体分裂,减缓低氧诱导的PASMCs 增殖。Zhao等[21]也发现,慢性缺氧导致 PASMCs 线粒体分裂和细胞增殖,乙醛脱氢酶2 通过下调4-羟基壬烯醛/HIF-1/DRP1 信号通路,减少PASMCs 线粒体分裂和细胞增殖,从而减轻PH。Shen 等[20]的研究则提示,通过抑制DRP1还可以减少内皮细胞线粒体分裂,降低PAECs 的增殖和迁移能力,从而减轻PH。此外,Dai等[22]发现,高氧诱导的支气管肺发育不良新生大鼠模型,在进入正常氧浓度环境后,肺动脉的阻力明显增加,而抑制DRP1表达则可以改善这类大鼠的肺血管发育,降低肺动脉压力。上述研究表明,通过抑制DRP1,减少线粒体分裂,可以抑制PASMCs 和PAECs的异常增殖,有望成为PH治疗的潜在靶点。
3.2 DRP1 适配体 DRP1 由细胞浆内转移至线粒体外膜的过程需要其适配体的协同,这些适配体包括线粒体分裂蛋白1(fission 1,FIS1)、MFF、MiD49和MiD51。FIS1 目前认为不是线粒体分裂所必需的成分[11,23]。近期,有研究对 DRP1 适配体在 PH 发生中的作用进行了探索。研究显示,无论是在PH 患者,还是PH 大鼠模型的肺动脉内膜、中层MiD49 和MiD51 的 表 达 均 明 显 升 高 ,PH 患 者 PASMCs 中MiD49和MiD51表达也明显升高。通过RNA 干扰降低MiD49 或MiD51 表达,可以逆转脉高压患者PASMCs 线粒体分裂,同时减少PASMCs 增殖,增加PASMCs 凋亡,从而减缓PH 的发展。他们同时发现MiD49和MiD51均是 microRNA-34a-3p 的靶基因,上调microRNA-34a-3p 可以取得相似的效果。而下调DRP1 的另外两个适配体FIS1 和MFF,则无法抑制PASMCs 线粒体分裂,减少 PASMCs 增殖[24]。上述研究提示,抑制 DRP1 适配体 MiD49 和 MiD51,同样可以作为PH治疗的潜在靶点。
DRP1 适配体在PAECs 线粒体分裂中的作用尚未见报道。近期,有研究报道DRP1适配体在其他血管内皮细胞线粒体中的作用。Zhou 等[25]发现,线粒体分裂增加与心肌缺血再灌注损伤有关。他们发现在缺氧复氧小鼠心脏微血管内皮细胞中FIS1、MiD49和MiD51 表达增加,线粒体分裂增加,导致血管内皮细胞功能障碍和凋亡增加,从而导致缺血再灌注损伤。此外,他们还发现,敲除MFF基因可以减少线粒体分裂,改善小鼠心脏微血管内皮细胞功能,减少心脏微血管内皮细胞死亡[26]。上述研究表明,FIS1、MFF、MiD49和MiD51均与心脏微血管内皮细胞线粒体分裂增加、细胞功能障碍有关。但在体循环血管内皮细胞中线粒体分裂导致细胞凋亡增加,而在PAECs 中线粒体分裂增加则表现为细胞增殖和凋亡抵抗,这是否与体肺循环血管内皮细胞生理特性上的差异有关,有待进一步研究证实。
3.3 线粒体融合相关蛋白 线粒体融合相关蛋白包括MFN1、MFN2和OPA1,MFN1和MFN2介导线粒体外膜的融合,OPA1则介导线粒体内膜的融合。抑制MFN1、MFN2 或OPA1 将会减少线粒体融合,促进线粒体分裂。因此,MFN1、MFN2 或 OPA1 也有可能成为干预 PH 的靶点。Ryan 等[27]发现,PH 患者和 PH大鼠的 PASMCs 中 MFN2 表达明显下调,而 MFN1 和OPA1 表达则无明显变化。在正常PASMCs 中下调MFN2,可增加线粒体分裂,促进PASMCs 增殖。PH患者和PH 大鼠的PASMCs 线粒体分裂增加,细胞增殖明显,而上调MFN2 则可以减少线粒体分裂,增加线粒体融合,从而减少PASMCs 增殖。在PH 大鼠中上调MFN2,可以增加大鼠的行走距离,降低肺血管阻力,降低肺动脉中层厚度,从而明显减轻PH。Fang等[28]也发现,缺氧诱导的PH大鼠,MFN2表达下调,PASMCs 增殖明显,凋亡减少,过表达MFN2 可以逆转缺氧诱导的PASMCs 增殖和凋亡减少。此外,Zhu等[29]和Lu等[30]的研究也均表明,MFN2表达下调与PASMCs 增殖和凋亡减少有关,而上调MFN2 表达则可通过增加线粒体融合,减少线粒体分裂,降低PASMCs 增殖,增加PASMCs 凋亡,从而维持两者之间的平衡。上述的研究提示,上调MFN2 可作为干预PH的潜在靶点。
但目前MFN1和OPA1对PASMCs线粒体分裂调控,及其在PH 中作用的研究相对较少。现有的证据显示MFN1 和OPA1 在PH 患者和动物模型的PASMCs表达没有明显的变化[27],但翻译后修饰可能在蛋白表达总量不变的情况,调整蛋白分子的功能[11]。因此,并不能除外 MFN1 和 OPA1 通过调控线粒体分裂参与PH 的发生。MFN1 和OPA1 对线粒体分裂的调控已在心肌细胞中得到验证。研究报道,缺氧引起心肌细胞中线粒体分裂增加,融合减少,导致心肌细胞坏死和功能障碍,而过表达OPA1可以逆转上述现象[31]。PH 大鼠右心室 MFN1 表达下调,通过上调microRNA-140 下调MFN1 的表达,可以加重右心室肥厚和增高右心室收缩压[32]。MFN1和OPA1在PASMCs 中是否具有相似的调控作用,需要专门针对MFN1或OPA1基因差异表达或蛋白翻译后修饰的研究来进一步阐明。
近期,有研究发现在肺微血管内皮细胞中下调MFN1、MFN2 和OPA1 均可引起线粒体分裂的增加,并且MFN2 还有维持肺微血管内皮细胞的屏障功能和抑制内皮细胞炎症起着重要的作用。但该研究中下调MFN2 并没有对肺微血管内皮细胞的增殖能力和凋亡造成明显的影响[33]。因此,尽管抑制MFN1、MFN2 和OPA1 可以促进肺微血管内皮细胞线粒体分裂,但对它们在肺血管内皮细胞增殖和凋亡中的影响,仍有待进一步的研究来阐明。
3.4 作用于线粒体动力代谢相关蛋白的翻译后修饰 翻译后修饰对于蛋白分子发挥其生物学功能起着重要的作用,比如磷酸化、泛素化等。研究表明,HMGB1(high-mobility group box 1)可通过激活细胞外信号调节激酶1/2(extracellular signal-regulated kinases 1/2,ERK1/2)而促进DRP1 磷酸化(但并不改变DRP1 总体表达水平),引起PASMCs 线粒体分裂增加,促进 PASMCs 诱导 PH 的形成,提示 ERK1/2 信号通路有可能参与了PH 的发生[15]。亦有研究显示,PH 患者 PASMCs 中 PINK1(PTEN-induced kinase 1)和蛋白激酶A(protein kinase A,PKA)可磷酸化MFN2 第 442 位丝氨酸,导致 MFN2 降解增加;抑制PINK1 或PKA 可增加MFN2 表达,减少线粒体分裂,从而抑制 PASMCs 增殖,诱导 PASMCs 凋亡[34]。因此,抑制MFN2 磷酸化信号通路也有可能参与了PH的调控。上述研究表明,除直接干预线粒体动力代谢相关蛋白的基因转录外,翻译后修饰也将是调节PASMCs线粒体分裂、增殖和凋亡的重要形式。线粒体动力代谢相关蛋白翻译后修饰对于PAECs线粒体分裂调控,及其在PH 发病中的作用目前尚无相关研究。相似的证据来自心脏微血管内皮细胞:核受体亚家族 4 A组成员 1(nuclear receptor subfamily 4,group A,member 1,NR4A1)通过激活酪蛋白激酶2α(casein kinase 2α,CK2α)而磷酸化MFF,从而促进线粒体分裂,加重缺氧复氧后心脏微血管内皮细胞的损伤[25]。但在PAECs 是否同样如此,有待进一步研究证实。
尽管线粒体动力代谢相关蛋白翻译后修饰已在PASMCs线粒体分裂、增殖和凋亡的调控中显示出重要的作用,但翻译后修饰繁多,且涉及众多的信号通路。翻译后修饰包括磷酸化、乙酰化、泛素化、类泛素化、棕榈酰化、蛋白质巯基亚硝基化和O-乙酰氨基葡萄糖修饰。磷酸化修饰涉及ERK1/2、糖原合成酶激酶3β、PKA、蛋白激酶B 和CaMKII(Ca²⁺/calmodulin-dependent protein kinase II)等信号通路。乙酰化修 饰 涉 及 SIRT (silent information regulator)和MARCH5(membrane-associated ring finger 5)等信号通路。泛素化修饰涉及parkin 和MARCH5 等信号通路。类泛素化修饰则与MAPL(mitochondria-associated protein ligase)信号通路有关。棕榈酰化修饰主要与 ZDHHC13(zinc finger DHHC-type palmitoyltransferase 13)信号通路有关[11,35]。此外,OPA1 的功能还与其蛋白的长度有关,在线粒体蛋白酶OMA1或YME1L 作用下,长片段OPA1 被分解成短片段OPA1,长片段OPA1 促进线粒体融合,而短片段OPA1 则促进线粒体分裂[36-37]。因此,线粒体动力代谢相关蛋白翻译后修饰是一个十分复杂的调控网络,且涉及的信号通路作用的蛋白底物广泛,不具备特异性,作为PH 潜在治疗靶点的可靠性与安全性仍需要进一步的研究证实(图1)。
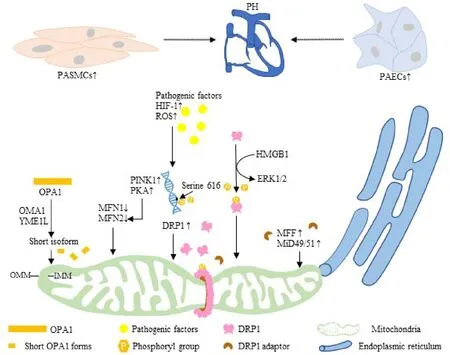
Figure 1. Potential target on regulation of mitochondrial fission to alleviate pulmonary hypertension(PH). PASMCs:pulmonary artery smooth muscle cells;PAECs:pulmonary artery endothelial cells;OMM:outer mitochondrial membrane;IMM:inner mitochondrial membrane;OPA1:optic atrophy 1;HIF-1:hypoxia-inducible factor-1;ROS:reactive oxygen species;MFN:mitofusin;PINK1:PTEN-induced kinase 1;PKA:protein kinase A;DRP1:dynamin-related protein 1;HMGB1:high-mobility group box 1;ERK1/2:extracellular signal-regulated kinases 1/2;MFF:mitochondrial fission factor;MiD49/51:mitochondrial dynamics protein of 49/51 kD.图1 肺动脉高压中线粒体分裂的调控靶点
4 总结
现有的研究证据表明,线粒体分裂增加促进PASMCs增殖和凋亡抵抗,同时也促进PAECs的增殖和迁移。以抑制线粒体分裂为靶点的干预方式,如抑制 DRP1、MiD49 和 MiD51 及上调 MFN2 在动物模型中均展现出良好减轻PH 的效果,为今后更有效治疗PH 提供了广阔的前景。但有如下问题亟待解决:(1)上述线粒体代谢相关蛋白在所有哺乳动物细胞中均起着调控作用,如何特异性地在PASMCs 和PAECs 中进行线粒体分裂抑制?(2)尽管线粒体分裂增加与PAECs 的增殖和凋亡抵抗有关,但目前仅有DRP1的相关证据,对于DRP1适配体、线粒体融合相关蛋白以及上述蛋白翻译后修饰在PAECs线粒体分裂、增殖和凋亡中的作用,仍有待阐明。(3)何种程度的线粒体分裂抑制是适度的?研究表明,完全敲除小鼠DRP1,导致心肌细胞坏死,出现致命性的扩张型心肌病[38]。因此,如何适度抑制线粒体分裂,将成为以抑制线粒体分裂为靶点治疗PH 最为关键的问题。上述问题也有待今后研究进一步探索。
