药用植物萹蓄叶绿体基因组特征与系统进化分析
2022-05-06胡赛文丁怡宁毕光耀李翠翠窦利军李贺敏
胡赛文,丁怡宁,毕光耀,李翠翠,苏 春,窦利军,李贺敏,夏 至*
• 药材与资源•
药用植物萹蓄叶绿体基因组特征与系统进化分析
胡赛文1,丁怡宁1,毕光耀1,李翠翠1,苏 春2,窦利军3,李贺敏1,夏 至1*
1.河南农业大学农学院,河南 郑州 450002 2.西北农林科技大学生命科学院,陕西 杨凌 712100 3.河南省信阳生态环境监测中心,河南 信阳 464000
以药用植物萹蓄为材料,利用高通量技术测定基因组DNA序列,对叶绿体基因组进行组装和序列分析,为进一步开展药用植物萹蓄的群体遗传学和遗传多样性研究奠定基础。利用华大MGISEQ-2000PE150测序平台,双末端测序策略对其全基因组DNA建库测序,测定萹蓄的基因组DNA序列,用NOVO Plasty组装叶绿体基因组,采用最大似然法(maximum likelihood,ML)构建系统进化树。萹蓄叶绿体基因组全长为163 461 bp,GC值37.5%,具1个典型的四分区域结构,包括1个大单拷贝区(large single copy region,LSC)、1对反向重复区(inverted repeats,IR)和1个短单拷贝区(small single copy,SSC),序列长度分别为88 023、31 066、13 306 bp。萹蓄的叶绿体基因组共有130个基因,其中编码蛋白基因、rRNA基因与tRNA基因的数量分别为83、8、37个。系统进化分析表明,萹蓄与木蓼属的额河木蓼构成一单系分支,具有100%支持率。萹蓄隶属于蓼科的蓼属,与木蓼属的额河木蓼亲缘关系较近。萹蓄叶绿体基因组的组装、序列特征、间隔区的筛选及系统进化分析为后续开展群体遗传学和遗传多样性研究奠定基础。
萹蓄;蓼属;叶绿体全基因组;组装;系统发育
药用植物萹蓄L.隶属于蓼科(Polygonaceae)蓼属L.,为一年生或多年生草本,主要分布在我国的河南、浙江、山东、河北、吉林等地[1-2]。萹蓄始载于《神农本草经》, 全草皆可入药[3],临床多用于治疗泌尿系统感染、肾炎、细菌性痢疾、非胰岛素依赖性糖尿病、结石等疾病[4-6]。《中国药典》2020年版规定萹蓄有效成分为杨梅苷(含量不得少于0.030%)[3]。此外,中药材萹蓄还含有丰富的化学成分,主要包括黄酮类、酚酸类、苯丙素类等[7]。现代药理学研究表明萹蓄具有利尿、降压、抑菌、抗氧化[8]和保肝等诸多的药理作用,临床应用十分广泛。目前,有关药用植物萹蓄的研究主要集中在化学成分分析和药理学研究等方面,该植物叶绿体基因组组装、基因组特征和系统进化分析等未见报道。
叶绿体不仅是绿色植物光合作用的重要场所、也是绿色植物最重要的细胞器之一。它利用太阳能和二氧化碳转换成化学能与氧气,维持地球生态平衡[9]、参与环境信号的响应,在逆境响应中也起着重要作用[10]。叶绿体具有一整套用于光合作用、能量代谢、蛋白质合成及氮、硫同化相关的基因,分布在大小为120~180 kb的环状基因组上,具有结构保守,母性遗传等特点[11-12]。叶绿体基因组一般为闭环双链DNA结构,陆生植物的叶绿体基因组结构通常由1个大单拷贝区域(large single copy,LSC)、1个短单拷贝区域(small single copy,SSC)和2个反向重复区域(inverted repeat,IR)组成[13]。叶绿体基因组拥有相对独立的基因组和遗传序列,并且不像核基因组一般有着复杂的重复序列,其基因序列保守,间隔区变异位点丰富,适宜的进化速率[14]能够为植物不同等级的亲缘关系,系统进化关系及遗传多样性研究提供较为可靠的信息[15]。
近年来,随着测序技术的不断改进,测序平台的不断升级,一系列组装和注释软件如plasmidSPAdes[16]、NOVOPlasty[17]、GetOrganelle[18]等的开发与更新,多种重要的药用植物叶绿体基因组已完成测序和分析,如丹参Bunge[19]、人参C.A.Mey[20]、三七(Burkill) F.H.Chen ex C.Chow & W.G.Huang[21]、粗茎秦艽Duthie ex Burk[22]、曼陀罗L.[23]、五味子(Turcz.) Baill[24]、铁皮石斛Kimura et Migo[25]和三叶崖爬藤Diels & Gilg[26]等。本研究以药用植物萹蓄为材料,利用高通量测序方法测定萹蓄基因组DNA序列,并对该植物的叶绿体基因组进行组装和注释。进而分析萹蓄叶绿体基因组序列特征,IR边界特征,间隔区信息位点的特征,并对萹蓄及其近缘19种植物的叶绿体基因组序列进行系统发育分析,验证其在科级系统发育中的位置,为药用植物萹蓄的种质资源的鉴定、开发和利用提供一定的理论依据。
1 材料与方法
1.1 材料
萹蓄新鲜叶片釆集于河南省新乡市原阳县小吴庄村河南农业大学科教园区(35°6′32″N,113°56′34″E),由河南农业大学生命科学院李家美教授和农学院中药材系夏至教授鉴定为蓼科植物萹蓄L.,凭证标本保存于河南农业大学标本馆,凭证标本号为XZ-2020-11。叶片装入取样袋后带回实验室,用无菌水冲洗数次,晾干后置于−80 ℃冰箱备用。萹蓄及其近缘物种叶绿体基因组序列来源于NCBI数据库,实验材料详细信息见表1。
表1 植物样品来源
Table 1 Plant samples used in this study
物种来源GenBank登录号 Afrobrunni chiaerecta (Asch.) Hutch.NCBIMH286316 额河木蓼Atraphaxis irtyschensis Yang et HanNCBIMG878984 吉木乃沙拐枣Calligonum jeminaicum Z.M.MaoNCBIMN202608 泡果沙拐枣C.junceum FlourNCBIMK854997 金荞麦Fagopyrum dibotrys (D.Don) HaraNCBIMF491390 荞麦Fagopyrum esculentum Moench.NCBINC 010776 何首乌Fallopia multiflora Thunb.NCBIMK330002 Fallopia sachalinensis (F.Schmidt) RonseNCBINC 047446 Muehlenbeckia australis (Forst.f.) CausedNCBIMG604297 山蓼Oxyria digyna (L.) HillNCBIMN564931 中华山蓼O.sinensis Hemsl.NCBIKX774248 萹蓄Polygonum aviculare Linn.var.vegetum Ledeb.河南原阳MW044669 虎杖Reynoutria japonica Houtt.NCBIMT301955 波叶大黄Rheum franzenbachii Munt.NCBIMN564923 掌叶大黄Rheum palmatum L.NCBINC 027728 小大黄Rheum mpumilum L.NCBIMN564927 皱叶酸模Rumex crispus L.NCBIMN055629 羊蹄Rumex japonicus Houtt.NCBIMN720269 Symmeria paniculate Benth.NCBIMH286353 蓝花丹Plumbago auriculata Lam.NCBIMH286308
1.2 DNA的提取和高通量测序
采用北京天根生化植物DNA提取试剂盒(Tiangen Biotech Co.,中国)提取样品萹蓄新鲜叶片的总DNA,利用1%的琼脂糖凝胶电泳检测DNA完整性。样品送至华大生物科技公司(北京)后,使用NanoDrop2000微量分光光度计(Thermo Scientific,美国)检测总DNA的纯度和浓度。MGISEQ-2000 PE150测序平台测序,测序完成后,利用华大自主开发的过滤软件SOAPnuke过滤参数,过滤步骤为(1)过滤接头:测序read匹配上adapter序列的25%或者以上则删除整条read;(2)过滤低质量数据:如果测序read中质量值低于20的碱基占整条read的30%或者以上则删除整条read;(3)去N:如果测序read中N含量占整条read的1%或者以上,则删除整条read。(4)获得Clean reads。数据以FASTQ格式储存,用于后续的拼接和注释。
1.3 叶绿体基因组的拼接和注释
叶绿体基因组的拼接釆用NOVOPlasty[17]程序,插入片段大小设为150 bp。过滤后的reads用Geneious 11.0.3[27]拼接软件组装成重叠群,并对组装中的简并碱基,进行人工修正。利用Geneq-Annotation of Organellar(https://chlorobox.mpimp golm.mpg.de/geseq.html),结合NCBI上已报道的何首乌(GenBank登录号:MK330002)注释结果对萹蓄叶绿体全基因组进行基因注释,参数为默认值,最后进行手动调整。tRNA用ANAGORNV1.2.38(https://chlorobox.mpimpgolm.mpg.de/geseq.html)预测。注释完成后,使用Geneious11.0.3[27]生成GenBank格式的文件,并提交到GeneBank(https://www.ncbi.nlm.nih.gov /genbank/),登录号为MW044669。利用在线工具OGDRAW-DRAW Organelle Genome Maps(https:// chlorobox.mpimp-golm.mpg.de/OGDraw.html)绘制叶绿体结构图。
1.4 IR边界的收缩和扩张分析
IR区域在叶绿体基因中具有高度保守性,但是IR/SC边界区域仍存在细微的变化,IR边界的膨胀和收缩被认为是被子植物叶绿体全基因组大小变化的主要机制[28]。在植物进化过程中,IR/SC边界不同程度的扩张和收缩导致了边界和基因组长度的多样性[29]。本研究使用Geneioous 11.0.3[27]软件获得蓼科7个属7种植物叶绿体基因组的IRA/IRB、LSC和SSC和边界基因的序列长度,进行比较分析,探讨蓼科植物叶绿体基因组IR边界的收缩和扩张特征。使用Adobe illustrator软件绘制7种蓼科植物叶绿体基因组IR边界对比图。
1.5 叶绿体基因组基因间隔区信息位点分析
相比叶绿体基因编码区,叶绿体基因间隔区在近缘物种间往往具有更高的变异位点,通常被用来构建属间、属内种间物种系统进化发育关系。本研究基于蓼科已注释的11个属17种植物的叶绿体基因组序列特征,利用phylosuite vl.2.1[30]提取17个物种叶绿体基因组32个共有的间隔区,利用MAFFT[31]进行多重比对。统计这些间隔区的信息位点百分率,为下一步构建蓼科属级以下物种系统进化关系提供分子标记。
1.6 系统发育分析
从NCBI数据库选取已公开发布的19个蓼科物种叶绿体基因组数据(表1),同时以近缘的白花丹科(Plumbaginaceae)蓝花丹L.为外类群,利用phylosuite vl.2.1[30]软件基于MAFFT[31]进行多重比对。系统发育分析采用最大似然法(maximum likelihood,ML),利用CIPRES Science Gateway服务器(http://www.phylo.org/)中RaxML-HPC2 7.6.3软件(Stamatakis 2006)构建系统发育树。利用Bootstrap(BS)(1000次重复)检验各分支的支持率。系统发育树导出后利用FigTree version1.4.2查看。
2 结果与分析
2.1 萹蓄叶绿体基因组的结构
测序结果去除接头和低质量的数据后,共获得2.32 Gb原始数据,原始reads为15 471 394条,Q20达97.86%。组装和注释后得到萹蓄的叶绿体基因组。结果表明,萹蓄为共价闭合的双链环状分子(图1),全长为163 461 bp,GC值35.6%,是一个典型的4分区域结构,包括1个LSC、1对IR和1个SSC,它们的长度分别为88 023、31 066、13 306 bp。IR、LSC和SSC区域的GC值存在一定的差异,其中,IR区域的GC值最高(41.3%),LSC(35.5%)和SSC(32.5%)均较低,详细信息见表2。
2.2 萹蓄叶绿体基因组的组成和特点
萹蓄叶绿体基因组共包括128个基因,非重复基因113个,其中83个编码蛋白基因、8个rRNA基因与37个tRNA基因。LSC区包含的基因最多,包括60个蛋白编码基因和22个tRNA基因;SSC区包括1个tRNA基因(-)和11个蛋白编码基因;所有的rRNA基因、14个tRNA基因和12个蛋白编码基因在IR区(表2)。其中,蛋白质编码基因中与自我复制相关基因除rRNA基因和tRNA基因外(表3),还包括14个核糖体小亚基基因、10个核糖体大亚基基因和4个RNA聚合酶亚基基因;光合作用相关的基因有46个,包括12个NADH脱氢酶基因、5个光合系统I基因、15个光合系统II基因、6个细胞色素复合物编码基因、6个ATP合酶基因、1个二磷酸核酮糖羧化酶大亚基基因和1个依赖ATP的蛋白酶单元p基因;此外还有5个其他功能基因及6个未知功能基因。在tRNA中-、-、-、-、-、-和-各有2个拷贝;4个核糖体RNA均有2个拷贝,分别位于反向重复区IRA和IRB。核糖体蛋白大小亚基编码的基因中,、、和这3个基因均有2个拷贝,其余为1个拷贝。NADH脱氢酶亚基中的基因及未知功能蛋白基因和的拷贝数均为2。

LSC和SSC:大单拷贝区域、小单拷贝区域;IRA和IRB:2个反向重复区域;内圈深色部分:GC含量
表2 萹蓄叶绿体基因组碱基组成及特征
Table 2 Base composition and characteristic of chloroplast genome in P.aviculare
区域A/%T/%C/%G/%GC/%蛋白编码基因数tRNA基因数rRNA基因数基因总数长度/bp LSC31.632.918.317.335.560220 8288 023 SSC35.632.017.015.432.511 10 1213 306 IRA/IRB30.128.719.721.641.312148 3431 066 总计31.131.519.118.437.583378128163 461
因19、基因分别横跨LSC/IRb、SSC/IRb边界,故未统计在表2内
the19 andF genes across the boundaries of LSC/IRb and SSC/IRb, and they were not listed in table 2
表3 萹蓄叶绿体基因组编码的基因
Table 3 Encoded genes present in chloroplast genome of P.aviculare
基因分组基因名称数量 tRNAtrnH-GUG、trnK-UUUa、trnQ-UUG、trnS-GCU、trnG-UCCa、trnR-UCU、trnC-GCA、trnD-GUC、t rnY-GUA、trnE-UUC、trnT-GGU、trnS-UGA、trnG-GCC、trnfM-CAU、trnS-GGA、trnT-UGU、trnL-UAAa、trnF-GAA、trnV-UACa、trnM-CAU、trnW-CCA、trnP-UGG、trnI-CAUc、trnL-CAAc、trnV-GACc、trnI-GAUac、trnA-UGCac、trnR-ACGc、trnN-GUUc、trnL-UAG37 rRNArrn16c、rrn23c、rrn4.5c、rrn5c8 核糖体蛋白小亚基rps2、rps3、rps4、rps7c、rps8、rpsll、rpsl2ac、rpsl4、rpsl5、rpsl6a、rpsl8、rpsl914 核糖体蛋白大亚基rpl2ac、rpl14、rpl16a、rpl20、rpl22、rpl23、rpl32、rpl33、rpl3610 RNA聚合酶rpoA、rpoB、rpoCla、rpoC24 NADH脱氢酶亚基ndhAa、ndhBac、ndhC、ndhD、ndhE、ndhF、ndhG、ndhH、ndhI、ndhJ、ndhK12 光系统I亚基psaA、psaB、psaC、psaI、psaJ、5 光系统II亚基psbA、psbB、psbC、psbD、psbE、psbF、psbH、psbI、psbJ、psbK、psbL、psbM、psbN、psbT、psbZ15 细胞色素b/f复合物亚基petA、petB、petD、petG、petL、petN6 ATP合成酶亚基atpA、atpB、atpE、atpFa、atpH、atpI6 Rubisco大亚基rbcL1 蛋白酶clpPb1 成熟酶matK1 包裹膜蛋白cemA1 乙酰CoA梭化酶亚基accD1 细胞色素C合成酶ccsA1 翻译起始因子infA1 未知功能蛋白Ycf1c、ycf2c、ycf3b、ycf46 总计 130
a和b分别表示含有1个和2个内含子;c表示含有2个拷贝基因
a represent one intron, b represent two introns; c represents two copies of the gene
内含子在基因表达调控中发挥重要作用,萹蓄叶绿体基因组中有18个基因有内含子。其中,-、、、、、、、、、、、、、和各有1个内含子,而、、具2个内含子。matK基因位于基因内,整个编码区为内含子的一部分,存在序列共用现象;基因的3′端与基因的5′端,基因的3′端与基因的5′端、的3′端与的5′端重叠。
2.3 1R边界的特征
蓼科7个属7个物种叶绿体全基因组的IR-LSC和IR-SSC边界比较表明,蓼科各属物种边界具有高度的保守性,其LSC/IRb边界(JLB),SSC/IRb边界(JSB)、SSC/IRa边界(JSA)和LSC/IRa边界(JLA)的侧翼基因完全相同,但扩张程度存在一定的差异。蓼科7个物种在JLB边界全部位于rps19基因内,中华山蓼、掌叶大黄和何首乌中LSC和IRb区向rps19基因扩张的程度相同,均为171、108 bp。泡果沙拐枣、荞麦、萹蓄和羊蹄的LSC和IRb区向rps19的扩张程度变异较小,分别为174 bp和108 bp、176 bp和103 bp、172 bp和107 bp、169 bp和110 bp。萹蓄的JLB边界扩张范围,位于7个种之内。JSB边界扩张范围显示,中华山蓼、掌叶大黄、泡果沙拐枣、萹蓄与羊蹄的边界非常相似,均位于基因内部,基因在IRb和SSC区的扩张变异范围, 分别为95 bp和2134 bp,95 bp和2140 bp,19 bp和2225 bp,64 bp和2186 bp,62 bp和2182 bp。萹蓄的JLB边界扩张范围,位于7个种之内。但何首乌和荞麦的基因已全部扩张到SSC区内部,距JSB边界分别为53 bp和196 bp。JSA边界的扩张范围显示,其侧翼基因完全相同,均为基因(位于SSC区)和基因(位于IRa区)。其中7个物种的rps15基因和基因距JSA边界的碱基长度范围变异较大,分别9~267 bp和0~282 bp。萹蓄的JSA边界扩张范围,位于7个种之内。JLA边界扩张范围显示,其侧翼基因完全相同,均为(位于IRa区)和(位于LSC区)2个基因。其中7个物种的和2个基因距JLA边界碱基长度范围变异不大,分别163~207 bp和2~63 bp。萹蓄的JLA边界扩张范围,位于7个种之内。萹蓄及6个蓼科植物的叶绿体全基因组边界图见图2。

图2 萹蓄及6个蓼科植物的叶绿体全基因组边界图
2.4 基因组特征比较分析
由于虎杖和额河木蓼的叶绿体基因组注释不完全,除了叶绿体基因组全长比较分析后,IR区、SSC区和LSC区序列长度未做分析。叶绿体基因组特征比较分析见表4。萹蓄及蓼科18种近缘植物叶绿体基因组的序列长度范围介于158 851~170 974 bp,其中皱叶酸模的叶绿体全基因组的长度最短(158 851 bp),的叶绿体基因组最长(170 974 bp)。萹蓄的叶绿体基因组长度为163 461 bp,介于蓼科19个物种的叶绿体基因组长度范围之内。萹蓄及蓼科18种近缘植物叶绿体基因组GC含量的范围为37.1%~38.3%,其中的GC含量最低(37.1%),的GC含量最高(38.3%)。萹蓄叶绿体基因组的GC含量为37.5%,介于蓼科19个物种叶绿体基因组GC含量之内。利用Geneious 11.0.3分别得到萹蓄及蓼科18种近缘植物的IR区、LSC区和SSC区序列,结果表明,IR区的序列长度范围介于30 399~34 631 bp,皱叶酸模的IR区最短(30 399 bp),的IR区最长(34 631 bp)。萹蓄IR区长度为31 066 bp,介于蓼科19个物种IR区长度范围之内。LSC区的序列长度范围介于84 888~88 340 bp,其中荞麦LSC区的序列长度最短(84 888 bp),何首乌LSC区的序列长度最长(88 340 bp)。萹蓄的LSC区的序列长度为88 023 bp,介于蓼科19个物种LSC区长度范围之内。SSC区的序列长度范围介于12 657~13 881 bp。其中,皱叶酸模的SSC最短(12 657 bp),何首乌最长为(13 881 bp)。萹蓄的SSC区的序列长度为13 306 bp,介于蓼科19个物种SSC区长度范围之内。
表4 19种植物叶绿体基因组的特征
Table 4 Chloroplast genome characteristics of 19 plants
物种叶绿体基因组长度/bpGC值/%LSC长度/bpSSC长度/bpIR长度/bp Afrobrunnichia erecta170 97437.188 05813 65434 631 额和木蓼164 19237.5−−− 吉木乃沙拐枣162 53537.588 16013 31930 528 泡果沙拐枣162 45937.588 10713 41630 468 金荞麦159 91937.985 13413 30930 738 荞麦159 59938.084 88813 87530 412 何首乌163 77337.588 34013 88130 776 Fallopia sachalinensis163 48537.587 70313 56631 108 Muehlenbeckia australis163 48437.488 16713 48530 916 山蓼160 69837.585 74813 17230 889 中华山蓼160 40437.585 50113 13330 885 萹蓄163 46137.588 02313 30631 066 虎杖163 41037.5−−− 波叶大黄161 68837.486 94612 78430 979 掌叶大黄161 54137.386 51813 11130 956 小大黄161 74937.386 99712 81230 970 皱叶酸模158 85137.685 03912 65730 399 羊蹄159 29237.585 02813 00630 629 Symmeria paniculata162 50138.386 92013 43531 073
“−”代表GenBank注释缺失,不完全
“−”represent GenBank annotation missing
2.5 叶绿体基因组基因间隔区信息位点分析
基于蓼科11个属17种植物(虎杖和未统计在内)的32个叶绿体基因组间隔区信息序列特征统计表明(表5),在32个叶绿体基因间隔区中,变异位点百分率变化范围为2.2%~23.4%,最高的为psbK-psbI基因间隔区,其变异位点百分率为23.4%。超过20%有8个,分别为ndhE-ndhG、psaJ-rpl33、psbK-psbI、rpl33-rps18、rps16-trnQ-UUG、trnE-UUC-trnT- GGU、trnF-GAA-ndhJ、trnT-GGU-psbD。这些变异位点百分率较高的叶绿体基因间隔区,能提供足够多的信息位点,为蓼科属间和种间物种进化关系及分子鉴定提供较高的分辨率。
表5 蓼科植物17个物种的32个叶绿体基因间隔区矩阵位点信息
Table 5 Information of 32 chloroplast intergenic region in 17 species of Polygonaceae
基因间隔区信息位点/%非信息位点/%一致位点/%基因间隔区信息位点/%非信息位点/%一致位点/% accD-psaI18.610.770.7rps7-trnV-GAC 4.3 2.093.7 atpF-atpH15.0 7.977.1rps16-trnQ-UUG21.112.866.1 atpH-atpI18.611.569.9rrn4.5-rrn513.5 1.984.6 clpP-psbB16.1 7.376.6rrn5-trnR-ACG 8.2 2.889.0 ndhB-rps7 2.2 2.095.8trnC-GCA-petN17.011.072.0 ndhC-trnV-UAC12.410.377.3trnD-GUC-trnY-GUA19.2 7.673.2 ndhE-ndhG20.720.458.9trnE-UUC-trnT-GGU20.813.266.0 petA-psbJ16.2 8.974.9trnF-GAA-ndhJ20.1 9.370.6 petB-petD 9.9 8.881.3trnL-CAA-ndhB 6.9 2.890.3 petN-psbM19.3 9.271.5trnL-UAA-trnF-GAA19.5 6.873.7 psaJ-rpl3323.013.963.1trnN-GUU-ycf1 7.9 4.887.3 psbE-petL19.0 8.972.1trnP-UGG-psaJ15.414.969.7 psbK-psbI23.4 9.067.6trnQ-UUG-psbK13.0 9.977.1 psbM-trnD-GUC17.112.470.5trnR-ACG-trnN-GUU 6.0 3.190.9 rpl33-rps1820.216.563.3trnT-GGU-psbD20.911.367.8 rpoB-trnC-GCA19.312.168.6ycf2-trnL-CAA 3.7 2.693.7
2.6 系统发育分析
叶绿体基因组序列用进行多重比对后,矩阵长221 423 bp,其中信息位点20 235 bp(9.1%),非信息位点22 977 bp(10.4%),一致位点178 211 bp(80.5%),利用最大似然法和贝叶斯法构建萹蓄及18种蓼科植物叶绿体全基因组的系统发育树(图3)。系统发育树结果显示,蓼科植物除国外分布的2个属和,位于系统树基部位置。其余的取样类群可以分为3个主要分支,分支A仅包括荞麦属2个种构成单系分支,具有100%支持率。分支B包括3个属,大黄属、山蓼属、酸模属构成单系分支具有100%,分支B内这3个属的单系性都得到100%支持率。分支C包括6个属,虎杖属、木蓼属、沙拐枣属、、何首乌属、蓼属构成单系分支具有74%支持率。分支C内,沙拐枣属的单系性都得到100%支持率。萹蓄与额河木蓼聚在一支具有100%支持率。何首乌属与虎杖属聚在一支,具有100%支持率。
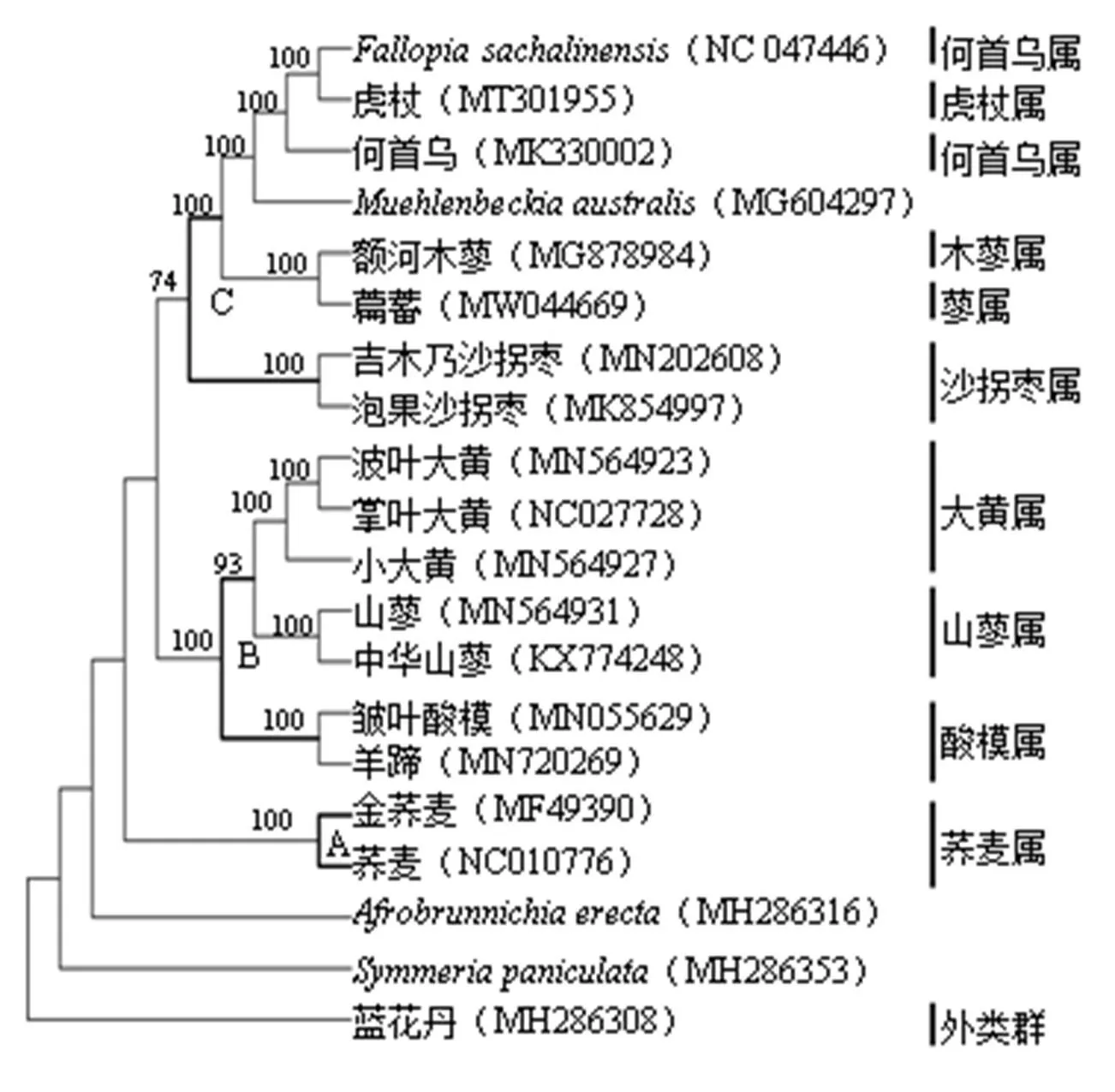
图3 基于叶绿体全基因组构建的系统发育树
3 讨论
萹蓄作为我国传统的药材之一,具有重要的药用和经济价值。本研究完成了药用植物萹蓄叶绿体基因组的测序、组装与注释。对比萹蓄与蓼科其他19个物种的叶绿体基因组分析结果表明,萹蓄叶绿体基因组具有典型的4分区域结构,包括1个LSC区、1对IR区和1个SSC区,其叶绿体基因组长度位于蓼科物种叶绿体基因组长度范围之内。被子植物中除部分列当科和兰科寄生植物的叶绿体基因组较短外,如[32](叶绿体基因组全长70 028 bp),天麻[33](叶绿体基因组全长35 304 bp)等,常见的叶绿体基因组大小为120~180 kb,共编码100~200个基因,包括70~80个蛋白质编码基因,30~32个tRNA基因,4个rRNA基因[34]。此外,除部分列当科和兰科寄生植物的叶绿体基因组部分基因发生丢失,具有较快的进化速率外,大多数植物的叶绿体基因组结构和序列相当保守,重复片段较少[33]。萹蓄与蓼科其他6个属植物的叶绿体基因组IR边界均具有高度的保守性,其LSC/IRb边界(JLB),SSC/IRb边界(JSB)、SSC/IRa边界(JSA)和LSC/IRa边界(JLA)的侧翼基因完全相同。且萹蓄的叶绿体基因组各边界的扩张范围,均位于蓼科物种叶绿体基因组IR边界的扩张范围之内。这表明在蓼科,萹蓄与其他物种的叶绿体基因组均具有较高保守性,适合用来解决属级以上分类等级的系统发育关系。
相对于叶绿体基因组编码区基因具有较高的保守性,叶绿体基因组的基因间隔区往往具有丰富的变异位点,本研究比较分析了蓼科17个物种的叶绿体基因组中32个基因间隔区,发现间隔区序列提供变异位点超过20%以上共计有8个,分别是ndhE-ndhG、psaJ-rpl33、psbK-psbI、rpl33-rps18、rps16-trnQ-UUG、trnE-UUC-trnT-GGU、trnF- GAA-ndhJ、trnT-GGU-psbD。这些基因间隔区在蓼科的属间和种间提供了丰富的信息位点。由于叶绿体基因组在大多数被子植物中为母系遗传,重组率低,核苷酸置换率适中[35],进一步结合双亲遗传的核基因片段联合分析,为蓼科植物属下大范围的物种鉴定,杂交起源,多倍体物种的形成和系统进化分析提供可靠的分子标记片段。
为进一步界定药用植物萹蓄在蓼科的系统位置,基于蓼科19个物种叶绿体基因组全长构建的系统发育树结果表明,除国外分布的2个种和,其余的17物种分别聚在3个不同分支内。萹蓄位于分支C内,与木蓼属的额河木蓼亲缘关系最近,二者构成一单系分支具有100%的支持率,形态特征也支持这2个种所在蓼属和木蓼属亲缘关系较近,二者均具有茎直立,花被片五枚,柱头片状,瘦果具翅等特征[1]。该分支内,虎杖属与何首乌属亲缘关系较近,二者构成一单系分支,具有100%支持率。分支B主要包括酸模族的3个属大黄属、酸模属和山蓼属,叶绿体全基因组的系统发育树支持酸模族的单系性,具有100%的支持率,形态学特征显示该族具有花被片4~6枚,雄蕊6~9枚,瘦果具3棱等特征[1]。大黄属,酸模属和山蓼属的各自单系性也得到100%的支持率。分支A主要由荞麦属的2个种构成,也组成一单系群,具有100%的支持率。本研究基于叶绿体基因组数据表明,萹蓄隶属于蓼科的蓼属,与木蓼属的额河木蓼亲缘关系较近。
近年来,随着新一代测序技术和生物信息学的快速发展,越来越多的药用植物物种的叶绿体基因组被报道。叶绿体基因组作为超级条形码或基于叶绿体基因测序分析筛选DNA片段显示出巨大的物种识别潜力,特别是在物种亲缘关系较近的物种间[36]。本研究首次报道了蓼科中药材萹蓄的叶绿体全基因组,综合分析蓼科药用植物如大黄、何首乌和虎杖等的叶绿体基因组序列,结构和特征,筛选一批叶绿体基因组的间隔区。这为蓼科药用植物的分子鉴定,种质资源保护,系统进化关系及遗传多样性研究奠定基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国科学院中国植物志编辑委员会.中国植物志(第17卷)[M].北京: 科学出版社, 1994: 126.
[2] 国家中医药管理局《中华本草》编委会.中华本草-2 [M].上海: 上海科学技术出版社, 1999: 639.
[3] 中国药典 [S].一部.2020: 348.
[4] 王桂芝, 胡海涛, 董鹏达, 等.中药萹蓄的研究现状 [J].黑龙江医药, 2010, 23(4): 614-616.
[5] 赵爱华, 赵勤实, 林中文, 等.萹蓄的化学成分研究 [J].天然产物研究与开发, 2002, 14(5): 29-32.
[6] 汤迎爽, 宋红儒, 杨丽甲.萹蓄的研究进展 [J].时珍国医国药, 2004, 15(1): 54.
[7] 李曼曼, 刘增辉, 王海燕, 等.萹蓄抑菌活性及化学成分研究 [J].天然产物研究与开发, 2014, 26(4): 526-530.
[8] 郑泽生, 杨爱梅, 张富禄, 等.萹蓄草提取物的抗氧化和抑菌活性研究 [J].中医药学报, 2019, 47(5): 55-58.
[9] Daniell H, Lin C S, Yu M,.Chloroplast genomes: Diversity, evolution, and applications in genetic engineering [J]., 2016, 17(1): 134.
[10] Pogson B J, Ganguly D, Albrecht-Borth V.Insights into chloroplast biogenesis and development [J]., 2015, 1847(9): 1017-1024.
[11] Twyford A D, Ness R W.Strategies for complete plastid genome sequencing [J]., 2017, 17(5): 858-868.
[12] Yu X Y, Zuo L H, Lu D D,.Comparative analysis of chloroplast genomes of fivespecies: Genome comparative and evolution analysis [J]., 2019, 689: 141-151.
[13] Zhang F J, Wang T, Shu X C,.Complete chloroplast genomes and comparative analyses of,, and(Amaryllidaceae) [J]., 2020, 21(16): 5729.
[14] 邢少辰, CLARKE JIHONG LIU.叶绿体基因组研究进展 [J].生物化学与生物物理进展, 2008, 35(1): 21-28.
[15] 张靖雯, 姜在民, 蔡靖.紫丁香与羽叶丁香叶绿体DNA提取方法研究 [J].西北林学院学报, 2018, 33(4): 95-99.
[16] Antipov D, Hartwick N, Shen M,.plasmidSPAdes: Assembling plasmids from whole genome sequencing data [J]., 2016, 32(22): 3380-3387.
[17] Dierckxsens N, Mardulyn P, Smits G.NOVOPlasty: De novo assembly of organelle genomes from whole genome data [J]., 2017, 45(4): e18.
[18] Jin J J, Yu W B, Yang J B,.GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes [J]., 2020, 21(1): 241.
[19] Qian J, Song J Y, Gao H H,.The complete chloroplast genome sequence of the medicinal plant[J]., 2013, 8(2): e57607.
[20] Zhao Y B, Yin J L, Guo H Y,.The complete chloroplast genome provides insight into the evolution and polymorphism of[J]., 2015, 5: 696.
[21] 宋菊, 龙月红, 林丽梅, 等.五加科植物叶绿体基因组结构与进化分析 [J].中草药, 2017, 48(24): 5070-5075.
[22] 董博然, 赵志礼, 倪梁红, 等.龙胆科叶绿体基因组结构特征及其药用植物鉴定意义 [J].中草药, 2020, 51(6): 1641-1649.
[23] Yang Y, Dang Y Y, Li Q,.Complete chloroplast genome sequence of poisonous and medicinal plant: Organizations and implications for genetic engineering [J]., 2014, 9(11): e110656.
[24] 郭豪杰, 刘久石, 罗丽, 等.五味子叶绿体基因组结构解析与比较分析 [J].中国科学: 生命科学, 2017, 47(7): 728-739.
[25] Biswal D, Konhar R, Debnath M,.Chloroplast genome sequence annotation of(Asparagales: Orchidaceae), an endangered medicinal orchid from northeast India [J]., 2017, 19: 1-8.
[26] 蒋明, 王军峰, 应梦豪, 等.三叶崖爬藤叶绿体基因组的组装与序列分析 [J].中草药, 2020, 51(2): 461-468.
[27] Kearse M, Moir R, Wilson A,.Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data [J]., 2012, 28(12): 1647-1649.
[28] Wang W B, Yu H, Wang J H,.The complete chloroplast genome sequences of the medicinal plant(Oleaceae) [J]., 2017, 18(11): 2288.
[29] Dong W L, Wang R N, Zhang N Y,.Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution [J]., 2018, 19(3): 716.
[30] Zhang D, Gao F L, Jakovlić I,.PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies [J]., 2020, 20(1): 348-355.
[31] Katoh K, Standley D M.MAFFT multiple sequence alignment software version 7: Improvements in performance and usability [J]., 2013, 30(4): 772-780.
[32] Wolfe K H, Morden C W, Palmer J D.Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant [J]., 1992, 89(22): 10648-10652.
[33] 马潇.兰科质体基因组组系统学研究 [D].北京: 中国科学院大学, 2018.
[34] 李岩, 吕光辉, 张雪妮, 等.十字花科植物叶绿体基因组结构及变异分析 [J].西北植物学报, 2017, 37(6): 1090-1101.
[35] Drouin G, Daoud H, Xia J N.Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants [J]., 2008, 49(3): 827-831.
[36] 姜汶君, 郭梦月, 庞晓慧.叶绿体基因组在药用植物鉴定及系统进化研究中的应用 [J].世界中医药, 2020, 15(5): 702-708.
Characterization and phylogenetic analysis of complete chloroplast genome of medicinal plant
HU Sai-wen1, DING Yi-ning1, BI Guang-yao1, LI Cui-cui1, SU Chun2, DOU Li-jun3, LI He-min1, XIA Zhi1
1.College of Agronomy, Henan Agricultural University, Zhengzhou 450002, China 2.College of Life Sciences, Northwest A&F University, Yangling 712100, China 3.Xinyang Environmental Monitoring Center of of Henan Province, Xinyang 464000, China
The complete chloroplast genome of medicinal plantsequenced by high-throughput technologies was assembled for the sequence analysis to provide evidence for its population genetics and diversity studies.DNA library was constructed forwith the paired-end strategy, and MGISEQ-2000PE150 was used to sequence DNA ofin Beijing Genomics Institute (China).The complete chloroplast genome was assembled using NOVO Plasty software, and sequence analysis was performed based on gene annotation results.Phylogenetic analyses were performed using Maximum-Likelihood (ML) methods.The complete chloroplast genome ofwas 163 461 bp in length with a GC content of 37.5%.The chloroplast genome exhibited a typical quadripartite structure, including a large single copy region (LSC), a pair of inverted repeats (IR), and a small single copy (SSC), and the sequence lengths were 88 023, 31 066 and 13 306 bp.The chloroplast genome harbored 130 genes, including 83 protein-coding genes, eight rRNA genes, and 37 tRNA genes.Phylogenetic analyses result indicated thatwas sister towith bootstrap value of 100%.Our result verified thatbelonged to the Polygonaceae, and it was closely related to.Sequence assembly, sequence features, intergenic region screening and phylogenetic analysis of medicinal plantchloroplast genome provide a basis for its future studies on both population genetics and genetic diversity.
L.;L.; chloroplast genome; assembly; phylogenetic analysis
R286.12
A
0253 - 2670(2022)09 - 2776 - 10
10.7501/j.issn.0253-2670.2022.09.021
2021-10-09
国家自然科学基金面上项目(31770370);河南省高等学校重点科研项目计划(18A360006)
胡赛文(1992—),女,硕士,研究方向为中药资源的分子鉴定。
通信作者:夏 至,教授,主要从事中药资源的分子鉴定及中药资源可持续利用研究。E-mail: xiazhiemail@126.com
[责任编辑 时圣明]
