基于二代测序技术的常染色体隐性遗传性多囊肾病家系胚胎植入前遗传学分析
2022-05-05何天文陈创奇丁红珂郑毅春尹爱华
何天文, 卢 建, 陈创奇, 刘 顿, 丁红珂, 刘 玲, 杜 丽, 郑毅春, 尹爱华
(1.广东省妇幼保健院医学遗传中心,广东 广州 511442;2.广东省妇幼保健院生殖中心,广东 广州 511442)
常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)是一种罕见的常染色体隐性遗传的单基因病,致病基因为多囊肾/多囊肝病变1(polycystic kidney and hepatic disease 1,PKHD1)基因[1]。ARPKD在临床上比较罕见,发病率仅为1/40 000~1/20 000,以肾脏和肝脏为主要累及器官,临床表型为双侧肾脏均匀对称性增大,并常伴有肝脾肿大、肝硬化及胆管扩张等症状,多数患儿于围产期或婴幼儿时即发病死亡[2-3]。ARPKD发病早、预后极差,目前临床尚无根治的办法,主要通过植入前遗传学检测(preimplantation genetic testing,PGT)或产前诊断预防ARPKD患儿的出生。近年来,二代测序[又称下一代测序(next generation sequencing,NGS)]技术飞速发展,测序成本不断降低,应用领域不断扩大。NGS的准确性好、精度高,目前已经成为胚胎PGT的主要方法之一,能检测染色体非整倍性、染色体结构异常和单基因遗传病[4-5]。国内外运用NGS技术对ARPKD家系进行PGT的报道较少,目前仅发布了《应用胚胎植入前遗传学检测技术阻断常染色体显性多囊肾病遗传的中国专家共识》[6]。为此,本研究对1个ARPKD家系进行PKHD1基因测序,在明确PKHD1基因致病突变的情况下,运用NGS技术对胚胎PKHD1基因突变位点进行直接测序,并构建胚胎PKHD1基因突变位点连锁单核苷酸多态性(single nucleotide polymorphism,SNP)位点单倍型,为胚胎PTG和ARPKD产前诊断提供参考。
1 材料和方法
1.1 研究对象
先证者父亲(Ⅰ1):35岁,表型正常,既往体健。先证者母亲(Ⅰ2):33岁,表型正常,既往体健,G2P1A2,2012年8月剖宫产一足月女(先证者);2016年12月孕8周胚胎停育,药物流产,未对绒毛进行相关基因检测。先证者(Ⅱ1):女,5岁,因血尿、蛋白尿以及B超显示肾脏有多发性囊状改变、肝脏囊性病变进行遗传性肾病相关基因检测,外院检测结果显示PKHD1基因c.5935G>A和c.10058T>G双重杂合突变,见图1。为了避免再次妊娠ARPKD患儿,该夫妇于2017年8月在广东省妇幼保健院医学遗传中心和生殖医学中心就诊咨询要求行PGT。向该夫妇充分解释PGT过程及相关风险后,该夫妇签署知情同意书并接受PGT。本研究经广东省妇幼保健院生殖医学伦理委员会批准。
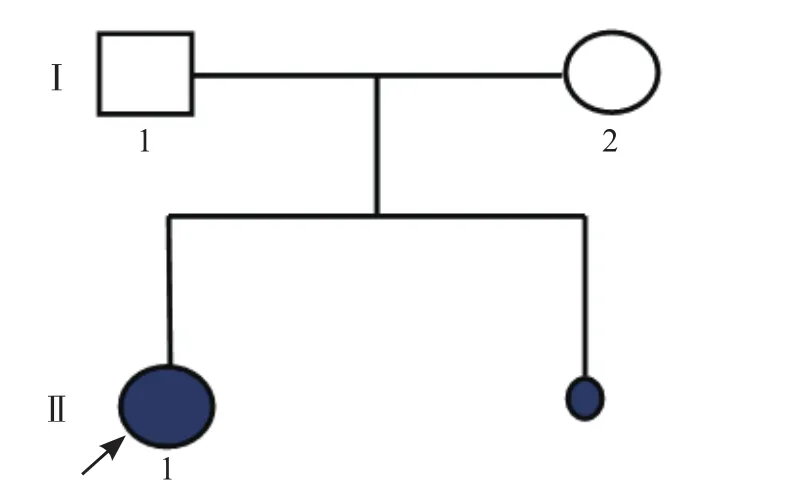
图1 ARPKD家系图
1.2 方法
1.2.1 样本采集及基因组DNA提取 采集先证者父母和先证者的静脉血各2 mL,乙二胺四乙酸抗凝,采用磁珠法血液基因组提取试剂盒[天根生化科技(北京)有限公司]提取外周血基因组DNA,严格按照试剂盒说明书操作。PGT后,先证者母亲于孕18~24周时在超声引导下行羊膜腔穿刺术,抽取羊水10 mL。采用Qiamp DNA Blood Mini Kit(德国Qigen公司)提取羊水基因组DNA,按照试剂盒说明书进行提取操作。
1.2.2 Sanger测序调查家系成员PKHD1基因突变情况 采用Oligo6软件(美国Oligo公司)设计针对PKHD1基因c.5935G>A和c.10058T>G突变位点的特异性聚合酶链反应(polymerase chain reaction,PCR)扩增引物。对扩增产物进行Sanger测序,测序仪器为ABI3730遗传分析仪(美国ABI公司)。采用SeqMan软件进行序列分析,观察PKHD1基因c.5935G>A和c.10058T>G的突变情况。
1.2.3 构建父母双方和先证者SNP单倍型 以PKHD1基因编码区为目标区域,在该基因上下游2M区域内选择120个高密度紧密连锁的SNP作为遗传连锁标记,在Ion AmpliSeq Designer网站(https://www.ampliseq.com)上设计多重PCR引物。扩增产物经纯化、建库、NGS后,选择有效SNP位点构建先证者父母和先证者的SNP单倍型,连锁分析后确定先证者父母携带PKHD1基因c.5935G>A和c.10058T>G突变的风险染色体。
1.2.4 胚胎培养及胚胎活检 常规促排卵取卵后,采用卵细胞质内单精子注射(intracytoplasmic sperm injection,ICSI)方式进行授精,受精后转入分裂期胚胎培养液继续培养,分别于培养第3、5、6天观察胚胎发育情况。胚胎发育第3天时用激光脉冲在透明带上切割出1个直径约20 μm的裂口,将胚胎转移至囊胚培养液中继续培养。胚胎发育第5天、第6天时观察到囊胚形成且脱出裂口滋养层细胞数为2~9个时进行活检。用活检针吸取脱出裂口的滋养层细胞,在磷酸盐缓冲液(phosphatebuffered saline,PBS)液滴中清洗后转入PCR反应管中。对活检的囊胚编号后进行玻璃化冷冻处理,待遗传学检测完成后再行解冻移植。
1.2.5 胚胎PGT 对活检获得的滋养层细胞进行基于等温多重置换扩增(multiple displacement amplification,MDA)技术的全基因组扩增。全基因组扩增产物通过多重PCR扩增PKHD1基因编码区和高密度紧密连锁的SNP。多重PCR扩增产物经纯化和建库后进行NGS。采用NGS对胚胎PKHD1基因突变位点进行直接测序,构建PKHD1基因突变连锁SNP单倍型并进行连锁分析。采用Sanger测序验证胚胎PKHD1基因突变位点的NGS结果。针对正常和杂合携带的胚胎进行低深度的染色体非整倍性筛查,以排除携带染色体拷贝数异常的胚胎。
1.2.6 胚胎移植和产前诊断 选择未检测到突变且发育良好的整倍体胚胎于冻融胚胎周期进行移植。胚胎移植14 d后查血人绒毛膜促性腺激素(human chorionic gonadotropin,hCG)确定是否妊娠;胚胎移植4周后行经阴道B超确定是否临床妊娠。孕18~24周时行羊膜腔穿刺,进行染色体核型分型和PKHD1基因c.5935G>A和c.10058T>G突变产前诊断,以验证PGT结果。
2 结果
2.1 ARPKD家系成员携带PKHD1基因c.5935G>A和c.10058T>G.突变情况
Sanger测序结果显示,ARPKD家系成员中,父亲携带PKHD1基因c.5935G>A,为杂合子;母亲携带PKHD1基因c.10058T>G,为杂合子;先证者携带PKHD1基因c.5935G>A和c.10058T>G双重杂合突变,分别遗传自父亲和母亲。见图2。

图2 先证者及其父母PKHD1基因c.5935G>A和c.10058T>G突变Sanger测序图
2.2 先证者父母和先证者的SNP单倍型
以PKHD1基因编码区为目标区域,在上下游2M区域内选择120个高密度紧密连锁的SNP位点作为遗传连锁标记。经过多重PCR和NGS检测后,选择69个有效SNP位点构建先证者父母和先证者的SNP单倍型,确定先证者父母携带PKHD1基因c.5935G>A和c.10058T>G突变的风险染色体。见表1。

表1 先证者父母有效SNP位点数和分布
2.3 植入前遗传学诊断
培养第5天观察到5个胚胎达到活检标准,即对囊胚进行活检和胚胎玻璃化冷冻保存,将活检产物标记为D501、D502、D503、D504和D505,每个样本活检细胞数为2~9个。采用NGS对胚胎PKHD1基因突变位点进行直接测序、胚胎单倍型连锁分析,同时采用Sanger测序进行验证。结果显示,5个胚胎中,D501和D502未检测到突变,D503和D505携带PKHD1基因c.5935G>A突变(杂合子),D504携带PKHD1基因c.10058T>G突变(杂合子)。见表2、图3。
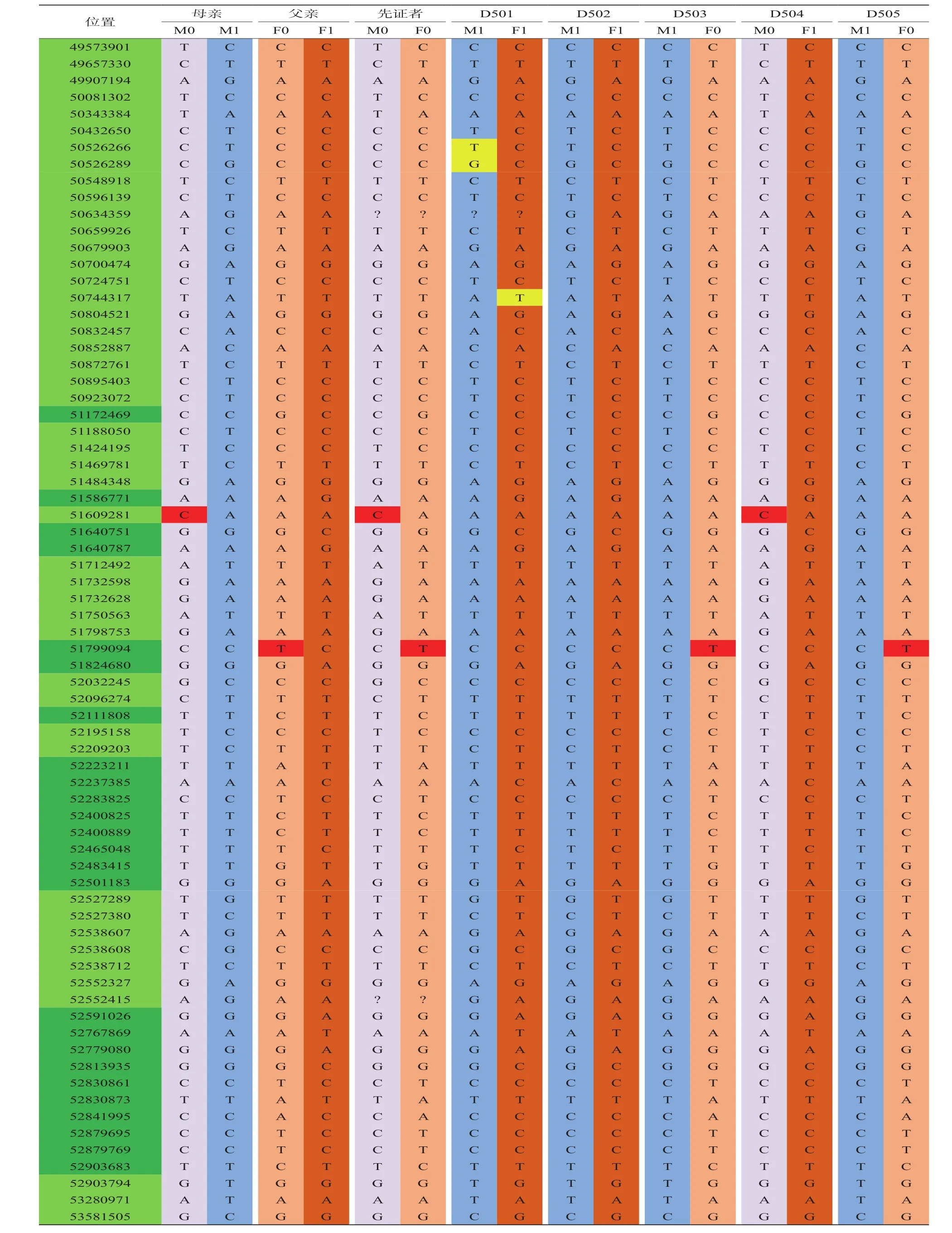
图3 胚胎单倍型连锁分析结果

表2 5个胚胎PKHD1基因c.472C>T突变NGS和Sanger测序结果
对5个胚胎进行低深度染色体非整倍体筛查,结果显示,D501、D503和D505为整倍体胚胎,D502和D504为非整倍体胚胎。见表3。
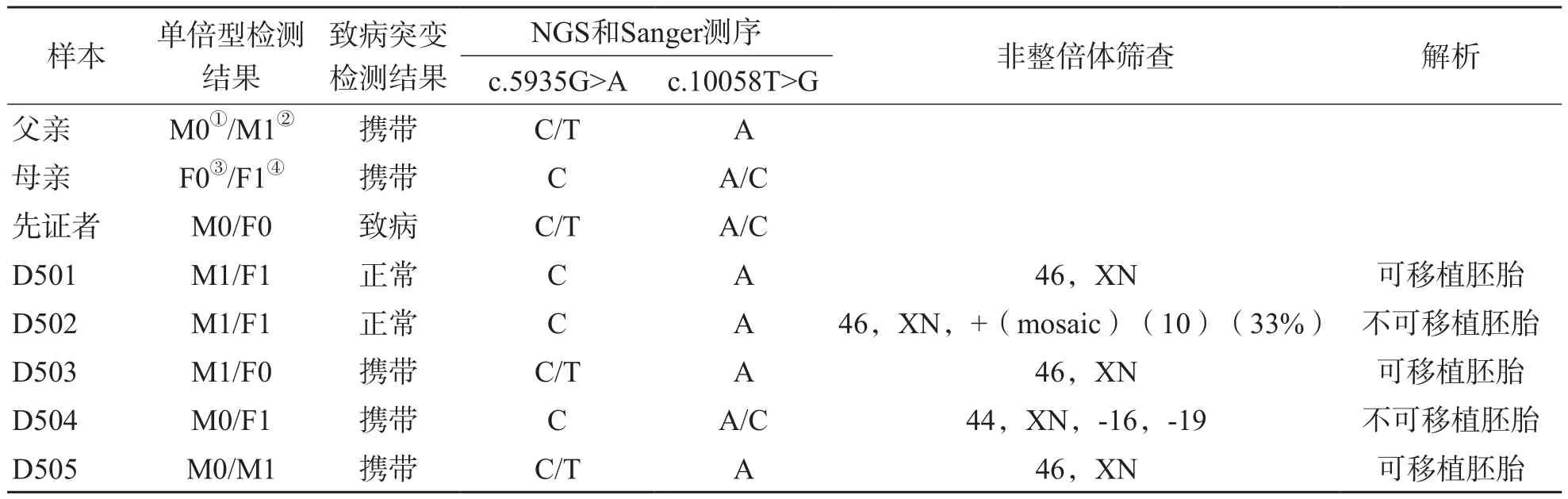
表3 胚胎PKHD1基因c.5935G>A和c.10058T>G突变PGT结果
2.4 等位基因脱扣率(allele drop-out,ADO)分析
5个活检胚胎的PKHD1基因致病突变位点c.5935G>A(chr6:51799094)和c.10058T>G(chr6:51609281)未发生脱扣。根据5个活检胚胎的2个致病突变位点和69个SNP位点的脱扣等位基因数得出复合ADO率为0.81%。
2.5 胚胎移植及产前诊断
囊泡活检后,在第3个月经周期行囊胚解冻移植。参考PGT结果选择遗传学检测结果为未检测到突变且发育良好的整倍体胚胎(D501)进行移植。移植14 d后检测外周血hCG为阳性,移植50 d后经阴道超声检查见单个孕囊,大小为34 mm×22 mm,胚芽长9 mm,可见心管搏动。孕18周经羊膜腔穿刺产前诊断结果显示,胎儿染色体核型分型无异常,未检测到PKHD1基因c.5935G>A和c.10058T>G突变(图4),与PGT结果一致。孕38周顺产一正常婴儿,健康状况良好。
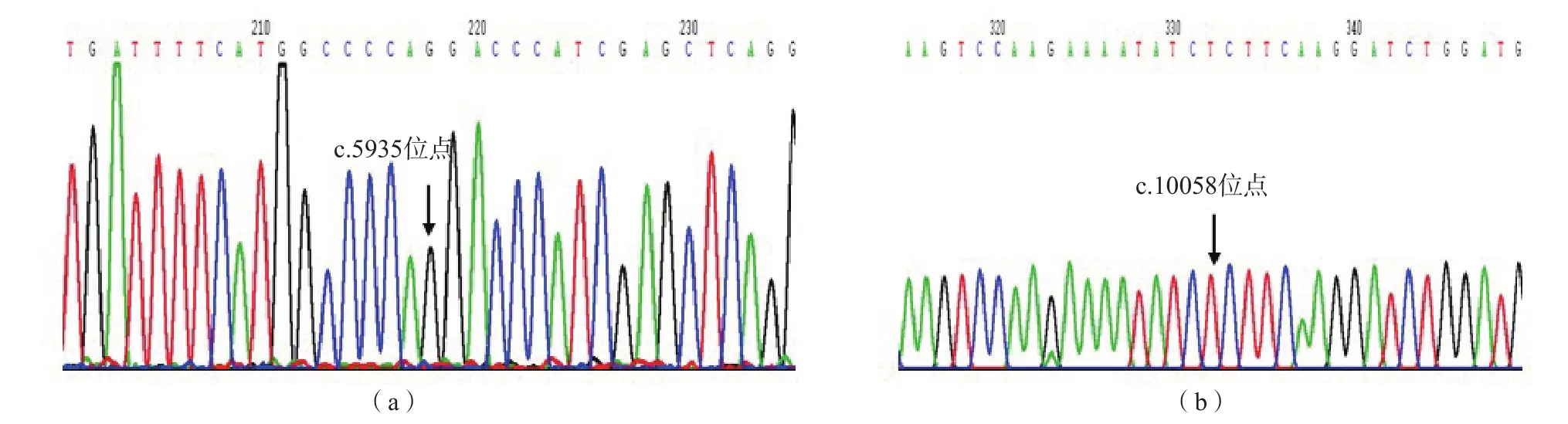
图4 胎儿PKHD1基因c.5935和c.10058位点的Sanger测序
3 讨论
ARPKD是一种罕见的常染色体隐性遗传的单基因病,致病基因为PKHD1基因。PKHD1基因是目前所知人类ARPKD的唯一致病基因,定位于人类染色体6p21,约为472 kb,包含67个外显子,编码1个由4 074个氨基酸组成的单次跨膜受体样蛋白——纤维素蛋白[7-8]。PKHD1基因以编码区的点突变致病为主,非编码区突变、大片段缺失、重复或重排只是致病原因的极少数。目前,基因变异数据库(http://www.humgen.rwth-aachen.de)中报道的PKHD1突变种类已达748种,其中约60%为错义突变,40%为截短突变(无义突变、剪切突变和移码突变)[9],其中c.107C>T(p.Thr36Met)和c.9689delA(p.Asp3230ValfsX9)是相对常见的突变,约占20%[10-11]。PKHD1基因发生突变会导致该蛋白结构或功能异常,从而影响肾脏集合系统及肝脏胆管系统发育及成熟。ARPKD以肾脏和肝脏为主要受累器官,临床表型为双侧肾脏均匀对称性增大,并常伴有肝脾肿大、肝硬化及胆管扩张等症状,多数患儿于围产期或婴幼儿时即发病死亡。ARPKD与大多数单基因疾病一样,尚无有效的根治方法。携带PKHD1基因突变的夫妇每次生育下一代都有25%的概率患ARPKD,通过产前诊断虽然可以防止ARPKD患儿的出生,但是绒毛活检、羊水穿刺、脐带血穿刺等传统的介入性产前诊断技术具有创伤性,孕妇有一定的流产风险。如果通过传统的介入性产前诊断确诊胎儿为ARPKD,则携带PKHD1突变基因的夫妇就需要选择终止妊娠。因此,在胚胎植入前进行遗传学检测,并选择正常胚胎进行移植,可以避免妊娠ARPKD胚胎,从而阻断PKHD1基因突变在家系中的传递。同时还可以对胚胎进行低深度的染色体非整倍体筛查,以排除携带染色体拷贝数异常的胚胎,避免选择非整倍体胚胎而导致的流产。因此,PGT与传统的产前诊断相比具有明显的优势[11-12]。
由于每个胚胎活检所得细胞数很少,提取的DNA量达不到分子检测所需的水平,需要通过全基因组扩增的方法来扩增基因组DNA。基于MDA技术的全基因组扩增技术[13]不但克服了单细胞水平分子检测DNA模板量过少的问题,也极大地提高了检测结果的准确性和单细胞水平基因检测的可靠性,但全基因组扩增技术会增加检测结果扩增不均匀或等位基因脱扣的风险[14]。NGS将单碱基测序成本降至了最低,也给PGT带来了革命性的改变[15]。有学者应用基于微卫星连锁分析的方法进行多囊肾病的PGT[16],相比之下,通过NGS完成植入前检测的时间周期更短、更高效。与传统的PGT技术,如荧光原位杂交和比较基因组杂交等相比,NGS具有明显优势,目前已逐渐成为PGT的主要检测方法[17]。本研究以PKHD1基因编码区为目标区域,在上下游2M区域内选择120个高密度紧密连锁的SNP作为遗传连锁标记,选择了69个有效SNP位点构建先证者父母的单倍型,确定其携带PKHD1基因c.5935G>A和c.10058T>G突变的风险染色体,减少了基因重组或全基因组扩增脱扣导致的检测结果错误。本研究统计了5个活检胚胎的2个致病突变位点和69个SNP位点的脱扣等位基因数,得出复合ADO率为0.81%。
综上所述,本研究成功应用NGS技术对ARPKD家系进行PGT,阻断了该单基因病在该家系中的再次发生,避免了选择非整倍体胚胎而导致的流产问题,是ARPKD出生缺陷的有效预防方法。
