豇豆中啶虫脒基体标准物质的研制
2022-05-01殷柯柯杨亚琴
刘 燕 殷柯柯 李 博 庞 泉 杨亚琴
(1. 河南省产品质量监督检验院,河南 郑州 450047;2. 河南省计量科学研究院,河南 郑州 450047;3. 天津市食品安全检测技术研究院,天津 300000;4. 河南省农业科学院农业质量标准与检测技术研究所,河南 郑州 450003)
啶虫脒(acetamiprid)又名乙虫脒、吡虫清,属于氯化烟碱类化合物,现常作为氨基甲酸酯、有机磷类农药替代品用于农业病虫害的控制[1]。2020—2021年,广东[2]、上海[3]、福州[4]、海南等地农产品市场监督抽检中不断有啶虫脒超标出现,实验室农产品日常监测工作中豇豆中啶虫脒检出率也较高。豇豆是花果同期作物,菜农在豇豆收获期施用农药控制花期病虫害容易导致农药安全间隔期无法保证,造成农残超标。啶虫脒是广谱高效杀虫剂[5],当被大剂量施用于农作物时也会造成农残超标。
基体标准物质[6-8]是具有实际样品特性并具有足够均匀性和稳定性特性值的物质或者材料,由于其目标化合物比基体更接近真实检测样品,能有效降低检测过程中的基体效应,保障检测结果准确性[9-19],被广泛应用于实验室质量控制工作。截至2021年,中国基体标准物质有36种,涉及水、土壤、水产、污水污泥、茶叶、浓缩苹果汁等类别。然而果蔬农产品基质水分含量高、易腐烂变质,基体标准物质常规制备以液体、果蔬浆添加稳定剂、增稠剂、防腐剂为主,技术成本高、稳定性不易保持,而且成品不方便贮藏运输。传统高温烘干、喷雾干燥工艺制备的样品复溶品质差、农残添加物易分解。冷冻干燥技术[20-21]生产的果蔬冻干粉能克服以上工艺技术缺陷,采用该技术制备的标准物质复水性好,复溶后的产品能极大程度保留原产品特性并且水分含量易于控制,方便贮藏和运输,且均匀性和稳定性良好。研究拟以豇豆为基体,制备啶虫脒豇豆基体标准物质,旨在为加强农产品质量安全控制、评估农残膳食风险水平、制修订食品安全标准工作提供依据。
1 材料与方法
1.1 材料与试剂
豇豆:无农药喷洒,农科院有机试验田;
农药啶虫脒溶液标准物质GBW(E)081861:标准值1 000.0 μg/mL,扩展不确定度9 μg/mL (k=2),农业部环境质量监督检验检测中心;
甲醇、乙腈:色谱纯,霍尼韦尔贸易(上海)有限公司;
丙酮:色谱纯,烟台市双双化工有限公司;
甲酸、乙酸铵:色谱级,天津市科密欧化学试剂有限公司;
陶瓷均质子(Agilent5982-9313)、萃取盐提取包(Agilent Bond Elut EN5982-5650)、吸附剂硫酸镁净化管(Agilent5982-5056):安捷伦科技有限公司。
1.2 仪器与设备
液相色谱质谱联用仪:Waters TQS型,美国Waters公司;
冷冻干燥机:SCIENTZ-12ND型,宁波新芝公司;
电子分析天平:XA205DV型,瑞士Mettler Toledo公司;
涡旋混合器:IKA MS3型,德国IKA公司;
冷冻离心机:CT 14RD TECHCOMP型,北京天美科学仪器有限公司;
24位氮吹仪:DC-24型,上海安普实验科技股份有限公司;
打碎磨粉机:800Y型,中国永康市铂欧五金制品有限公司;
去离子水发生器:Milli-Q型,美国Millipore公司;
三维混合机:SYH-1型,江苏天力干燥工程有限公司;
钴源工业辐射装置:Co60,河南省科学院同位素研究所辐照中心。
1.3 试验方法
1.3.1 基体标准物质制备 取空白阴性豇豆样品25 kg粉碎至软糊状,放入冻干机托盘预冷冻后冻干处理。出仓打粉过40目筛,得到空白基质标准物质。取2 L大烧杯,将设计量的啶虫脒农药用丙酮稀释至一定体积,分3次倒入装有2.0 kg空白基体标准物质的大烧杯中不断搅拌,液体没过空白基体标准物质,继续搅拌3 min,加玻璃表面皿放置于通风橱内等待溶剂自然挥发至接近物料上表面。将物料于真空度0.001 Pa下冻干48 h,粉碎过40目筛,于温度20 ℃、湿度35%下低速混匀36 h,水分保持在5%以下。混匀的样品由检验员在干燥仓内手动分装至5 mL螺旋口棕色储液瓶,并装入铝箔袋抽真空封口包装,每单元5 g/瓶。集中采用钴源工业辐射装置辐照处理,辐照剂量11 kGy,辐照时间1 h,于超低温冰箱贮藏备用。
1.3.2 基体标准物质啶虫脒含量测定 根据文献[22-23]并修改。
(1) 样品处理:称取1.000 g样品(精确至0.001 g)于50 mL塑料离心管中,加入9 mL超纯水涡旋混匀使其复溶,静置20 min。参照GB 23200.121—2021中的QuEChERS前处理方法进行提取和净化。继续加入10 mL 乙腈、陶瓷均质子剧烈振荡1 min,加入萃取盐包继续剧烈振荡1 min,4 ℃、5 000 r/min离心5 min,取6 mL 上清液加入含有净化吸附剂和硫酸镁的15 mL净化管中涡旋混匀1 min,5 000 r/min离心5 min,取上清液过Captiva RC有机滤膜,上LC-MS/MS测试,采用基体标准溶液外标法定量检测。以1.000 g阴性样品(精确至0.001 g)为对照,得空白基质标准溶液。
(2) 液相色谱条件:色谱柱为HSS T3 2.1 mm×100 mm×1.8 μm;流动相A为5 mmol/L乙酸铵+0.1%甲酸;流动相B为乙腈;流速0.3 mL/min;色谱柱温度40 ℃;进样量2 μL,按表1进行梯度洗脱。

表1 液相色谱梯度洗脱条件Tabe l Conditions of gradient elutionin HPLC
(3) 质谱条件:电离模式为ESI+;多反应MRM监测;去溶剂温度500 ℃,去溶剂气流速1 000 L/Hr,雾化气压力0.7 MPa,离子源温度150 ℃,气帘流速150 L/Hr,毛细管电压2.5 kV,锥孔电压30 V;啶虫脒多反应监测母离子(m/z)223,定量子离子(m/z)126,定性子离子(m/z)99。
1.3.3 方法学验证 称取1.000 g阴性样品(精确至0.001 g)于50 mL塑料离心管中,精密加入适量啶虫脒标准溶液,使其含量约为0.25,0.50 mg/kg,混匀后按1.3.2中样品处理方法制得样品加标回收测定液,供LC-MS/MS上机测试。
1.3.4 均匀性检验 按JJF 1343—2012执行。采用单因素方差分析(F检验)对检测数据进行统计分析[24]。
1.3.5 稳定性检验 按JJF 1343—2012执行,采用同步稳定性评估方法以减少评估各时间点测量结果的离散型。考察-18 ℃贮藏条件下的长期稳定性和30 ℃传递过程的短期稳定性。
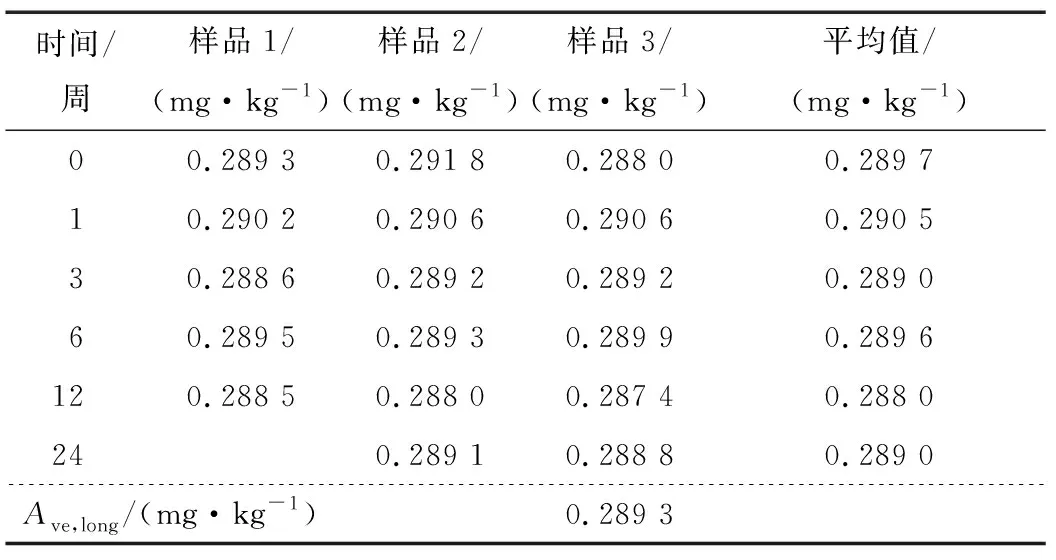
(1) 长期稳定性:随机抽样,采用先密后疏的原则选取时间间隔为0,1,3,6,12,24周,每个时间点抽取3个样品,每个样品平行测定3次,并采用线性拟合方程模型进行统计分析。
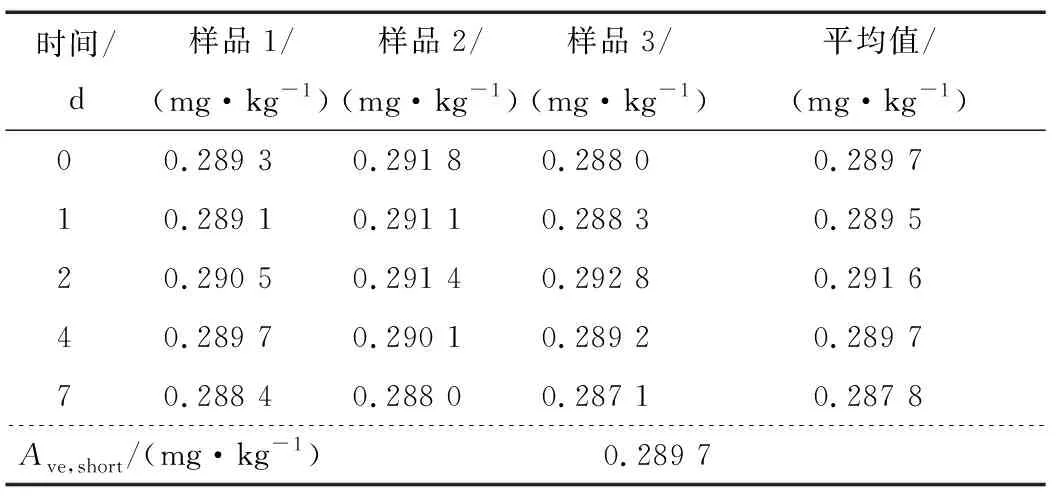
(2) 短期稳定性:采用t检验法,选取时间间隔为0,1,2,4,7 d,每个时间点抽取3个样品,每个样品平行测定3次取平均值。
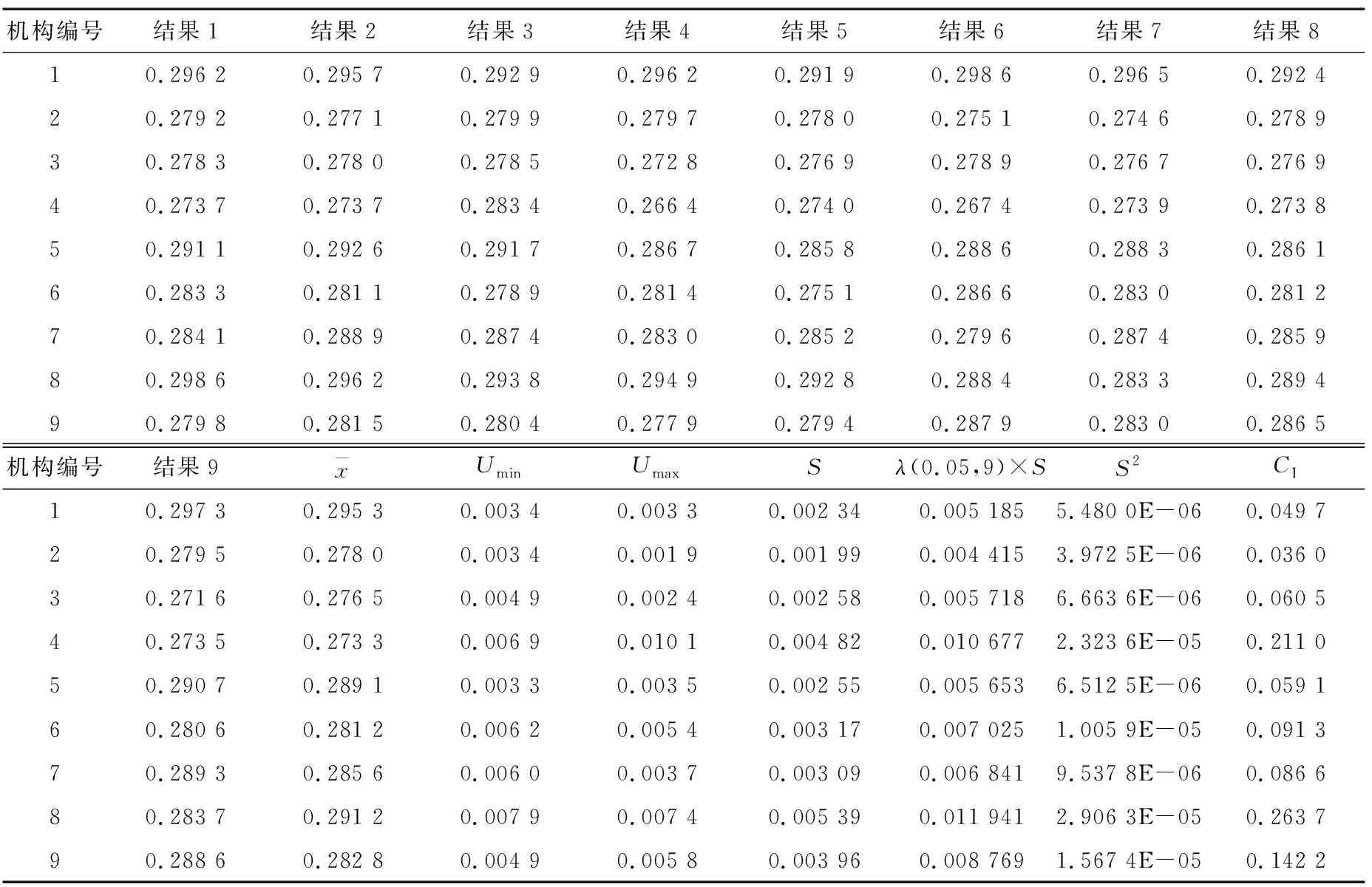
1.3.6 合作定值 采用9家通过资质认定的具有技术权威性的检验检测机构作为合作定值实验室,编号分别为1~9,每个实验室3个样品,每个样品平行测定3次,通过Grubbs和Cochran检验进行数据处理,剔除离可疑值和离群值,将剩余的数据取总平均值作为制备基体标准物质的特性量值。
(1) 格拉布斯(Grubbs)检验:按式(1)、式(2)对9家合作定值实验室内检测数据是否有显著差异进行验证评估。
Umin<λ(0.05,9)×s,
(1)
Umax<λ(0.05,9)×s,
(2)
式中:
Umin——各实验室内检测结果最小值与平均值的差值的绝对值;
Umax——各实验室内检测结果最大值与平均值的差值的绝对值;
s——各实验室内检测数据标准偏差。
查λ(0.05,9)=2.215,如果式(1)、式(2)同时成立,说明该实验室内数据无离群值。否则剔除离群值,并重新进行Grubbs检验直至最后剩余的数据均能通过该检验。
(2) 科克伦(Cochran)检验:按式(3)对9家合作定值实验室测定结果平均值是否等精度进行验证评估。
(3)
式中:

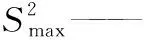
如果C
(3) 参与定值设备及规格型号:见表2。

表2 仪器品牌和型号Tabe 2 Instrument brand and model
1.3.7 基体标准物质不确定度评估 依据标准物质统计方法指南[25]规定,评估合作定值方法得到定值结果的不确定度,考察均匀性、稳定性、合作定值测定和B类不确定度共同的贡献。
(1) 均匀性标准不确定度ubb评估:通过F法检验均匀性良好的基体标准物质考察均匀性引入的标准不确定度。
(4)
式中:
S1——组间标准偏差;
S2——组内标准偏差。
(2) 稳定性标准不确定度us评估:采用趋势分析法,考察线性关系斜率随样品稳定性变化的变化值。该研究考察了长期稳定性引入的不确定度ults和短期稳定性引入的不确定度usts,稳定性不确定度按式(5)计算。
us=s(β1)×d,
(5)
式中:
ults——长期稳定性标准不确定度;
usts——短期稳定性标准不确定度;
s(β1)——β1的标准偏差;
d——保存期限。
(3) 合作定值测定引入A类标准不确定度ucharA评估:由9家检验检测机构采用同种方法对生产的基体标准物质定值,参与机构检测数据的总平均值作为特性值,合作定值测定引入的标准不确定度按式(6)计算。
(6)
式中:
S——各检验检测机构定值结果平均值的标准偏差;


m——合作定值机构数量。
(4) 合作定值引入B类标准不确定度ucharB评估:包含样品称量引入的相对标准不确定度urelB1、标准物质纯度引入的相对标准不确定度urelB2、体积引入的相对标准不确定度urelB3、实验室温度引入的相对标准不确定度urelB4和溶液移取引入的相对标准不确定度urelB5。
(7)
ucharB=urelB×x。
(8)
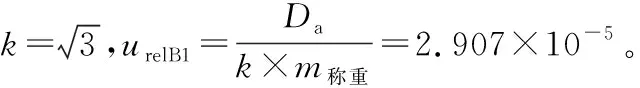
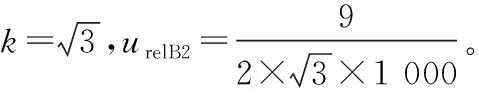

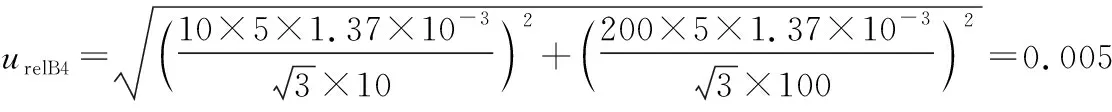

(5) 合成扩展标准不确定度:上述各不确定度分量合成标准不确定度uCRM,乘以包含因子k,即为基体标准物质的扩展不确定度UCRM。
(9)
2 结果与分析
2.1 方法学验证
通过添加不同含量水平的啶虫脒标准物质测定其加标回收率,由表3可知,回收率为99.5%~105.6%,相对标准偏差<2%,验证结果满足定值要求。
2.2 基体标准物质均匀性检验
随机从封装好的单元中抽取15个样品进行均匀性检测,每个样品测定3次,结果见表4。检验结果采用单因素方差分析法(F检验法)进行统计分析,F 表4 均匀性统计结果†Table 4 Statistical analysis results of homogeneity testing 2.3.1 短期稳定性 由表5可知,|β1| 表5 短期稳定性结果统计†Table 5 Statistical analysis results of short-term stability testing 2.3.2 长期稳定性 由表6可知,|β1| 表6 长期稳定性结果统计†Table 6 Statistical analysis results of long-term stability testing 对9家检验检测机构的测量数据首先采用Grubbs检验,查表得λ(0.05,9)= 2.215,考察各家实验室内检测数据的最小值残差Umin<λ(0.05,9)×s和最大值残差Umax<λ(0.05,9)×s,经过考察9家检验检测机构数据均通过Grubbs检验。继续采用科克伦法检验各家实验室平均值间是否等精度,查表得C(0.05,9,9)=0.266,计算得出C=0.263 7并且满足C 表7 9家实验室合作定值结果†Table 7 Cooprative fixed value of 9 laboratories 通过冻干技术、样品外源加标、Co60辐照灭菌相结合工艺,采用液相色谱—质谱串联法检测、基质标准溶液校正定量以及多家实验室合作定值的方式建立了一种豇豆中啶虫脒基体标准物质的制备方法。结果表明,该方法工艺简单,易于生产控制,制备的基体标准物质极大程度保留了基质样品本底特征,样品复原性、均匀性、稳定性均良好,通过9家实验室合作定值最终确定特性值为(0.283 7±0.008 5) mg/kg (k=2)。制备的产品可用于检测设备期间核查、新方法确认与验证、实验室间组织比对等实验室质量管理工作。 鉴于农残多残留基体标准物质有着更广泛的应用面,且考虑市场实际需求该研究同时进行了以豇豆为基体的啶虫脒、嘧菌酯、氯虫苯甲酰胺和噻虫嗪多残留协同研究。稳定性保持和精准定值是多残留标准物质制备的关键控制点,但该研究多残留稳定性控制尚未达到目标物协同正相关预期,下一步将继续加强多残留基体标准物质的保存工艺研究。农产品基体种类繁多,农残问题复杂,市场需求量大,农残基体标物设计上应以市场急需和突发情况为主,同时拓宽特性量值种类,提升定值技术水平。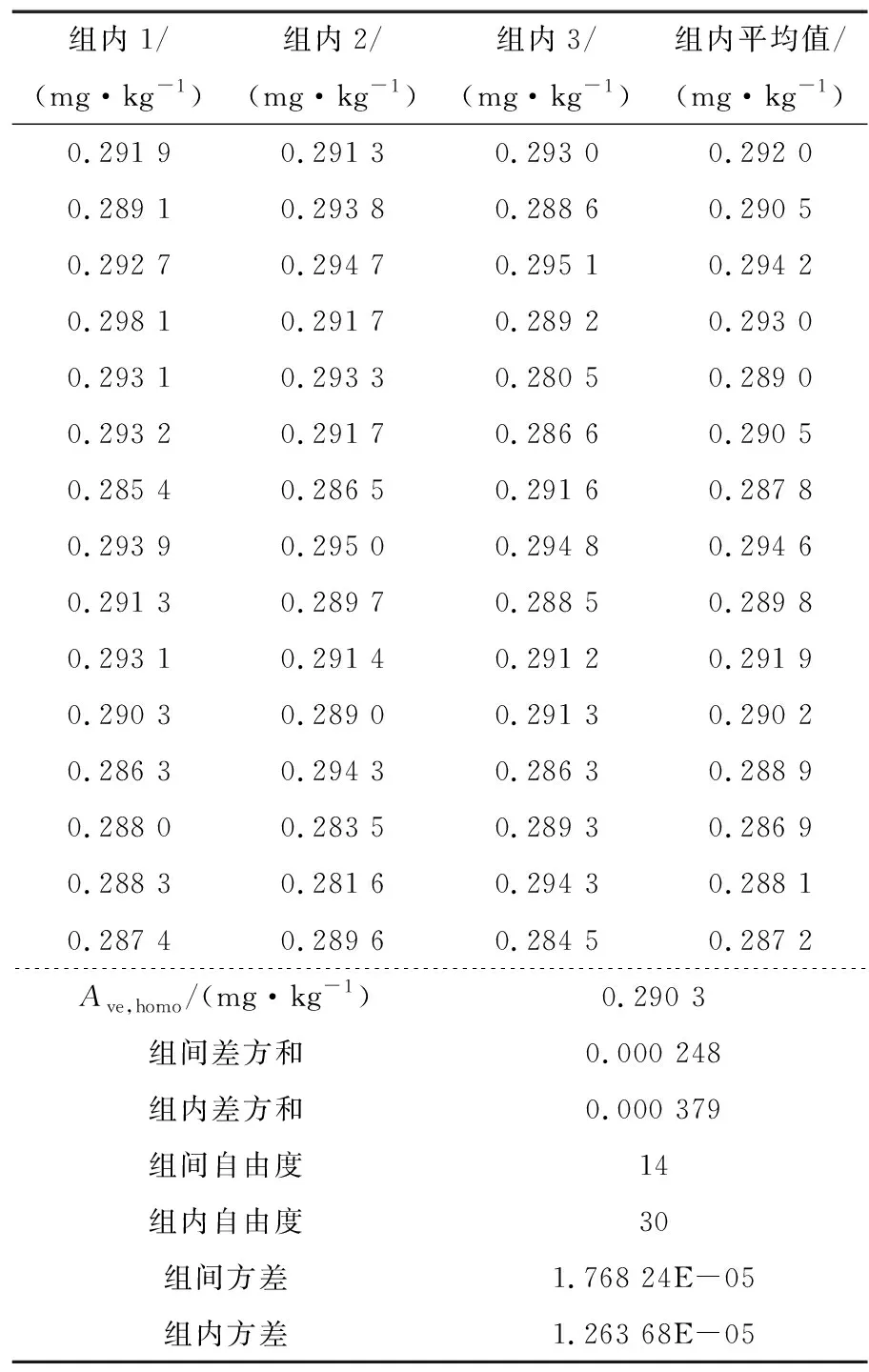
2.3 基体标准物质稳定性检验
2.4 基体标准物质合作定值
2.5 基体标准物质不确定度评估
3 结论
