The quasi-activity coefficients of non-electrolytes in aqueous solution with organic ions and its application on the phase splitting behaviors prediction for CO2 absorption
2022-04-27XiaomengZhaoXingyuLiChangjunLiuShanZhongHoufangLuHairongYueKuiMaLeiSongSiyangTangBinLiang
Xiaomeng Zhao ,Xingyu Li,2 ,Changjun Liu ,Shan Zhong ,Houfang Lu,2 ,Hairong Yue,2 ,Kui Ma,Lei Song,Siyang Tang,*,Bin Liang,2
1 Laboratory of Low-Carbon Technology and Chemical Reaction Engineering,School of Chemical Engineering,Sichuan University,Chengdu 610065,China
2 Institute of New Energy and Low-Carbon Technology,Sichuan University,Chengdu 610207,China
Keywords:CO2 absorption Polyamines Biphasic system Phase-splitting Salting-out effect
ABSTRACT CO2 capture with a low energy consumption is of important application significance for reducing CO2 emission.The phase-change absorbent developed in recent years shows its potential for low-energy CO2 capture.The unclear phase-splitting rule hinders the efficient development of CO2 phase-change absorbents.To predict phase-splitting behaviors of mono/poly-amine-organic solvent–water system with various concentrations,a quasi-activity coefficient was developed based on Debye &McAulay equation and some Density function theory descriptors.Six models based on Debye &McAulay equation were developed with different ion radius,descriptors or poly-amine-CO2 products.The phase-splitting boundary was drawn on the model with the best predictability.This quasi-activity coefficient would provide guidance for the phase-splitting systems development,especially for polyamines.
1.Introduction
Carbon dioxide(CO2)emission accompanied with human activities seriously affects the environment [1,2].Carbon capture,utilization and storage (CCUS) technology is regarded as an effective approach to alleviate climate challenge [3].CO2separation from fuel gas is an important part of CCUS.The chemical absorption is one of the most mature and common methods for CO2capture.The amine aqueous system has been applied for CO2absorption in industry since 1930s[4].However,the high regeneration energy required leads to the high power generation cost [5].Many researches focus on reducing the regeneration energy by the process optimization [5],catalytic regeneration [6,7] and new absorbents development [8,9].Phase-splitting system,as a new kind absorbent,is usually homogeneous before absorption but heterogeneous after absorption.The CO2absorption product dissolved in water would be condensed into one phase.A CO2lean phase and a CO2rich phase then could be observed.Energy consumption can be effectively reduced by regenerating CO2rich phase[10].The diethylenetriamine(DETA)-sulfolane system reduces the heat duty by 19%compared with monoethanolamine(MEA)aqueous system[11],while MEA-AMP (3-amino-1-propanol) system shows a 36%off of regeneration energy than MEA aqueous system[12].Comparing with amine aqueous systems,phase-splitting systems show the great potential for low-energy applications.
Abundant amine-based phase-splitting systems developed are classified into three types:primary/secondary amines-tertiary amine-water system [13,14],primary/secondary amines-organic solvent–water system [15,16] and primary/secondary amineorganic solvent system [17,18].The CO2chemisorption products may ionize as Eqs.(1)–(3).The product of Eq.(1) is regarded as the salting-out trigger [12,19].And Eq.(2) and Eq.(3) occur with ample water in the systems [20,21].For those aqueous systems,the ‘‘salting-out”effect between the electrolyte and nonelectrolytes was considered as main reason for phase-splitting[14,16,22,23].

To rapidly develop new phase-splitting absorbents,many approaches were carried out to quantitatively reveal the relationship between molecular structure and the absorption behaviors[23–26].Recently,new ion lgP models were developed for predicting phase-splitting behaviors of the amine-organic solvent–water system for CO2absorption [23].Although the models show good predictability,the models are still limited to mono-amine systems.Poly-amine systems have large absorption capacities and prone to occur phase splitting[27,28].Poly-amines are with more than one N atoms.It may form mono-carbamate [RNHCOO]-,bi-carbamate[R(NHCOO)2]2-,tri-carbamate [R(NHCOO)3]3-and so on after CO2absorption.A phase-splitting model broadly applicable to both mono-amine system and poly-amine system may provide more solutions to design new systems.However,due to the complexity of the poly-amines in CO2absorption,it is difficult to predict their phase-splitting behaviors.
In traditional inorganic salting-out researches,Setschenow constant (Eq.(4)) [29] and Debye &McAulay theory (Eq.(5)) [30,31]were developed to predict the salting-out performance.Setschenow equation defined as the solubility different of solute in ionaqueous system,is relative to the activity coefficient.The activity coefficient of non-electrolyte in Debye &McAulay equation was quantified according to the Gibbs free energy difference and Born polarization model.The solute is prone to salting out with the lnγ over 0.These two equations are suitable for describing the salting-out phenomenon caused by electrostatic force,and those could be applied for anions and cations with different charge and concentration.

In Eq.(4),S0is the solubility of non-electrolyte in pure water,S is the solubility in the presence of salt,γ is the activity coefficient of the non-electrolyte in the electrolyte solution,KSis Setschenow constants,c is the salt concentration [32].

In Eq.(5),z is the charge of ion,β is the dielectric decrement,ε is the static dielectric constant of the solvent,r±is the ionic radius,c is the salt concentration in aqueous solution,and k is the Boltzman constant.
The specific ion–solvent interactions of amine-organic solvent–water systems were investigated in our previous study [23].The chemisorption products of amine were anions [RNHCOO]-and cations [RNH3]+.The ions were dissociated in CO2-rich phase but associated in CO2-lean phase.According to the energy decomposition,the binding energy between ions and solvents are dominated by electrostatic force[33].Based on the analysis,a Debye&McAulay equation with only electrostatics considered may work in organic ions system for further amine-CO2absorption calculation[30,32,34].Most Debye &McAulay work focued on inorganic salts[35,36],and the key parameter β of organic ions was barely reported.Therefore,if a similar Debye &McAulay equation could be adjusted with some proper semi-empirical parameters,it is probably applied to predict the phase-splitting behavior of polyamine systems with different material proportion.
This work aims at developing a similar activity coefficient of the non-electrolyte in the organic electrolyte aqueous solution.It would be further applied in predicting the phase-splitting behaviors of CO2absorption by amine-organic solvent–water systems with different material proportion.The absorption products of the poly-amine system are experimentally detected.A similar Debye &McAulay equation with a quasi-activity coefficient (lnγ′)is developed and expressed with some density function theory(DFT) parameters.Six regression models of the organic-aqueous solution are developed based on 92 datasets.A phase-splitting prediction diagram with good predictability is then drawn and discussed.
2.DFT Calculation Details and Experimental Section
2.1.DFT calculation
For each amine-organic solvent–water system,the geometryoptimization of each ion (anion,cation) and the interaction of the ion–solvent were conducted at the functional level of M062X and the basis set of 6-311++G(d,p) with Gaussian software [37–39].The D3 version of Grimme’s empirical dispersion with the original D3 damping function [40] and the polarizable continuum model (PCM) using the integral equation formalism variant(IEFPCM)were added to accurately calculate the molecular properties.The geometry information was obtained with Multiwfn software [41].Detailed calculation methods were shown in SSection 1.1 (Supplementary Material).
The nuclear magnetic resonance (NMR) nuclear shielding results were carried out at the B3LYP/6-311++G(d,p) level,which has been proved with a good agreement with experiment values in DETA-water systems [42].
2.2.Experimental section
2.2.1.Materials
1,3-Diaminopropane(PDA)(AR>99.5%)and 1,4-butanediamine(BDA) (AR >98%) were purchased from Shanghai Adamas Reagen Co.,Ltd.,China.Methanol (AR >99.5%),ethanol (AR,>99.5%),1-propanol (AR >99.5%),2-propanol (AR,>99.5%),ethylene glycol(AR,99%) were purchased from Shanghai MACKLIN Biochemical Co.,Ltd.,China.CO2(>99%) and N2(>99%) were supplied by Chengdu Dongfeng Gas Co.,Ltd.,China.
2.2.2.CO2absorption
For detecting chemisorption products of the polyamines,the CO2absorption experiment was performed as shown in Fig.1.All experiments were conducted at 313 K with the amine of 2 mol.L-1and organic solvent of 3 mol.L-1.The mixed gas(CO2volume fraction of 15%)was injected to the isothermal reactor at a flow rate of 500 ml.min-1controlled by a two-channel mass flowmeter (MFC,Beijing Sevenstar Electronics Co.Ltd.,China).When the volume fraction shown in the CO2analyzer is consistent with preformulated CO2,the reaction was terminated.All phase-splitting behaviors were then observed after 24 h.NMR (JNM-ECZ400S/L1,JEOL Ltd.,Japan)was performed,and all acquired spectra was analyzed with Jeol Delta software.
3.Theory and the Calculation
3.1.The quasi-activity coefficient lnγ′
Two drawbacks limit the application of Debye&McAulay Equation.(1) only electrostatic effect is considered;(2) the lnγ′value depends on the dielectric decrement β,which could be expressed as the dielectric constant difference of the solute in water and non-electrolyte over the molecule number of solventThe dielectric constant of the aqueous solution changes with the variance of the salt and solvent [43,44].It is an ion specific property.If the presence of one ion can lower the dielectric constant of the aqueous solution,salting-out may occur in the system.The compositions of amine-organic solvent–water system is complex,including the CO2-amine chemisorption products (cations,anions and ion pairs),amine,organic solvent and water.It’s hard to measure the activity coefficient of the component after CO2absorption.
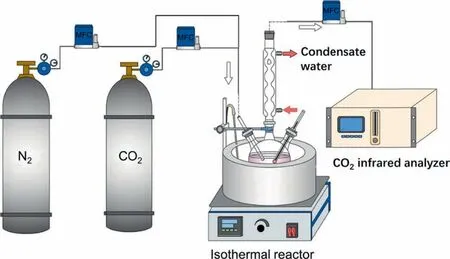
Fig.1.Experiment device for CO2 absorption.
A quasi-activity coefficient (lnγ′) is proposed according to Debye&McAulay equation.It is developed with some DFT parameters to describe the salting-out ability of non-electrolyte as shown in Eq.(6).In the traditional Debye &McAulay equation,the f(ε) is expressed as the dielectric decrement β over the static dielectric constant of the solvent ε.It could be partial expressed as ε0-ε.ε0is dielectric constant of pure water,and ε is the dielectric constant of organic solvents.Here,the f(ε)represents the dielectric constant influences.z is the charge of ion,r±is the ionic radius,c is the salt concentration in the aqueous solution,and k is the Boltzman constant,f(ε) is a function of the static dielectric constant of the solvent.b is an adjusted parameter to compare the modified Debye&McAulay equation to 0.

In this amine-organic solvent–water systems,the expression of f(ε) is the key for the lnγ′calculation.In our previous study,some descriptors of the organics or ions were proved highly revealing the interaction between solvents and the CO2chemisorption products.Then a parameter α describing organic solvents properties is introduced as Eq.(7).This parameter α is a synthetic parameter obtained from previous QSPR(quantitative structure property relationship)and DFT calculation.It can reflect geometric and electrostatic configuration of organic solvents and ions.The positive or negative correlations of the descriptors are determined according to previous study [23].This parameter α is designed for adjusting the function f(ε).Then a similar Debye &McAulay equation and a quasi-activity coefficient lnγ′are constructed for further research.

Vsis the volume of organic solvents,is the maximum electrostatic potential value on solvent molecular surface,(S/V)sis the surface/volume value of organic solvents.
All the descriptors in Eq.(7) were generated by the software of Multiwfn and Gaussian for describing the ions and the solvents properties.The descriptors of the ions and the solvents are listed in Section 1.2 of Supplementary Material.
3.2.The radius used for lnγ′ calculation
In the Debye&McAulay equation,the ionic radius is highly relative to the salting-out behavior.The organic ions are different from the inorganic ions.The Debye &McAulay equation based on Born polarization model assumes the ions as non-polarizable spheres.But organic anions and cations are non-spherical.Here,to improve model accuracy by comparing different forms of ionic radius,the anions and cations radius were calculated in four different ways as follows:
(1) r1,defined as Eq.(8).It is the sum of the distance from the farthest atom to the geometry center of anion or cation (d)and the van der Waals radius of the atoms(ratom)(Fig.2(a)).

(2) r2,defined as Eq.(9).It is the sum of r1and half of a modified parameter to describe the penetration of van der Waals surfaces(dpenetration)(Fig.2(b)).It reflects the interaction of the ions and solvents.

(3) r3,defined as Eq.(10).For the penetration of specific two molecular couldn’t accurately represent the real situation in solvents,the average penetration of the ions was taken into r3.Here,the average value of d and dpenetrationwere used.

(4) r4,defined as Eq.(11).The anion and cation volumes on the iso-surface of the electron density of 0.01 were used to calculate the ion radius (Fig.2 (c)).

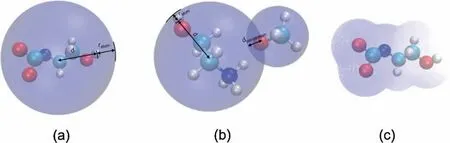
Fig.2.The calculation methods of ionic radius.
The average ionic radius was calculated as Eq.(12) for further lnγ′study.The x1and x2present the ratios of cations and anions,respectively.And all ionic radius (r1-r4) were listed in S-Section 1.3 of Supplementary Material.

3.3.Data set collection
Nighty-two amine-organic solvent–water systems were collected for this quasi-activity coefficient study.Eighty-two sets of data were collected from literatures,while ten sets of data were experimentally investigated in this work.Mono-amines (such as MEA,2-AP,3-AP,MAE,DIPA),bi-amines (such as BDA,PDA) and tri-amines (such as DETA) were included in this data set.Fortysix monophasic systems and forty-six biphasic systems were included (see Table 1).
3.4.lnγ′ model details
To establish proper lnγ′equation for amine-organic solvent–water systems,six models with different expression of ionic radius,f(ε) or CO2chemisorption product of polyamines were considered as shown in Table 2.
4.Results and Discussion
4.1.The CO2 chemisorption products of polyamines
Different DETA carbamates were considered in Fig.3.The calculated NMR spectrum of different DETA carbamates were listed in Table 3.Two signals (δ=39.356;δ=49.210) were confirmed in the molecular DETA (Fig.3(a)).The values of the chemical shift and the difference of chemical peaks were in agreement with the Ref.[42].However,the chemical shift of carbamates (δ=150–155) deviated from experimental value (δ=160–165) about 10.

Fig.3.The CO2 chemisorption products of DETA.(a)DETA;(b)the mono-carbamates of the primary amine;(c)the mono-carbamates of the secondary amine;(d)and(e),the di-carbamates;(f) the tri-carbamates.
The13C NMR chemical shifts of DETA-1-propanol-water system were as shown in Fig.4.The signals of δ=164.433,δ=164.492 and δ=164.525 could be ascribed to the primary amine carbamate.The signal of δ=163.994 and δ=163.928 belong to the secondary amine carbamate.The difference between the peaks of the primary carbamates (bC5,dC5,eC5,eC6,fC5 and fC6) was hard to distinguished.The chemical shifts of the secondary amine carbamates(cC7,dC7,fC7) were barely distinct with each other.However,some differences can be tell form C1 to C4 signals.The signal δ=49.283 and the signal δ=39.434 could present the peaks of aC1,aC4 and aC2,aC3.No bC3,bC4 or cC2 signal was observed.The signal δ=39.224 was the only signal smaller than aC1(aC4).Considered the calculated results,it could be ascribed to eC1 and eC4.The signals δ=39.799 and δ=39.926 could be ascribed to cC4 and dC1,while the signals δ=40.107 and δ=40.218 could be ascribed to fC1.The signal δ=49.066 indicated the presence of dC2.The signal δ=47.876 and δ=48.479 might indicate the presence of dC3,eC2,eC3,fC2 and fC3.
In summary,no mono-carbamate was found at the terminal of the CO2absorption according to13C NMR results.Bi-carbamate(d,e) and tri-carbamate (f) signals could be found,and di-carbamate formed from two primary amine groups(e)was the major product of CO2chemisorption.Distinguished from amine-water systems,no(δ=160.71) existed.According to the results,[DETA(COO)2]2-and [DETA(COO)3]3-should be both considered in the model building.
4.2.lnγ′ model regression
Different lnγ′models were shown in Table 4,and detailed data of all the models were in Section 1.4 of Supplementary Material.

Table 1 The data collected from literatures and experiments

Table 2 Different lnγ′ models

Table 3 The calculated NMR spectrums of DETA and different DETA carbamates
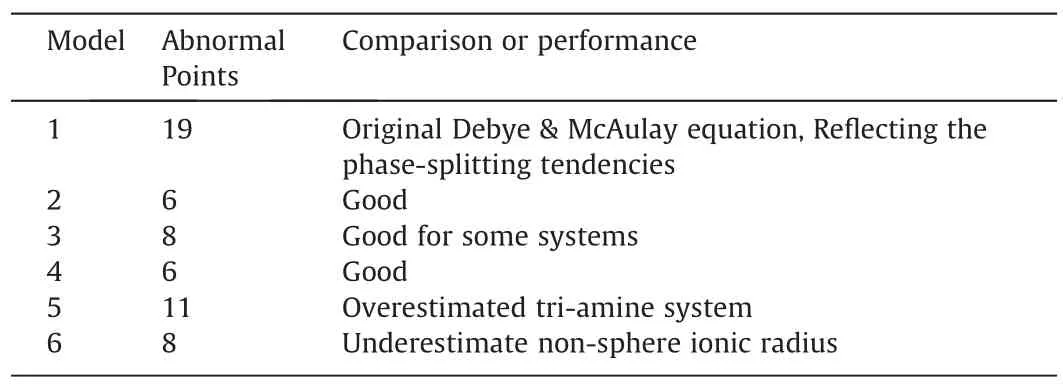
Table 4 The Prediction performances of different models
Model 1 was calculated according to the original Debye &McAulay equation.With no parameter α adjustment,Model 1 only reflected phase-splitting tendency.In this model,the organic solvent property was reflected in the dielectric constants,and ion property was reflected in the ionic radius.In a real aqueoussolution system,solvent volume,H-bonds and solvent geometric configuration influenced the salting-out behaviors.Therefore,the solvent MeCN with a high polarity but no H-Donor (No.9 MEAMeCN-water system) was prone to salting-out.And the sulfolane systems with a high polarity,a large volume but no H-Donor were the outlies in Model 1.
Model 2 introduced the parameter α to describe the organic solvent properties,and the predictability has been greatly improved with only six abnormal points.It showed a clear boundary between bi-phase systems and mono-phase systems.Except three abnormal points of mono-amine systems,two abnormal points of bi-amine systems and one abnormal point of tri-amine systems,good prediction performance of poly-amine systems was shown in Model 2.As discussed in the previous study[23],the descriptors(Vs,ESPs+,(S/V)s) did improve the quality of the regression model.
Model 2,Model 3,Model 4 and Model 6 took different ionic radius into the regression.Model 3,Model 4 and Model 6 considered the geometric configurations and the interaction between ions and solvents.Model 3 took r2as ionic radius,which reflecting the specific ion–solvent interactions.If the ions have strong polarity,it was prone to penetrate and ion radius reduced.Eight abnormal points existed in Model 3.Due to the instability of the coordination,the penetration of specific two molecular couldn’t accurately present the real situation in the solvents.Model 3 cannot describe the system stably.Since the ionic radius of the sameions may differ in the system,the average penetrations were taken as r3and used for Model 4 regression.Six outliers existed in Model 4,which were exactly same with Model 2.However,the average distance of abnormal points to boundary line of Model 2(0.724)was smaller than that of Model 4(0.750).Model 2 showed the better predictabilities.
Model 6 took r4as ionic radius with only ion volume considered.If the shape of the ion varies from the spherical shape,r4would be quite different with r1,r2and r3.The outliers in Model 6 indicated that this model might underestimated ionic radius with ions of non-sphere shape.
Comparing to Model 2 with the bi-carbamate as the product,Model 5 considered the tri-carbamate as the phase-splitting trigger.Model 5 had twelve abnormal points.The lnγ′of tri-amine systems were overestimated.The presence of tri-carbamates was minimal among all the models.The results also reflected that tricarbamate may not be the phase-splitting trigger.It is consistent with the13C NMR results.
4.3.lnγ′ models for predicting the phase-splitting behaviors
As discussed before,Model 2 (i-carbamate as the phasesplitting trigger,r1as the radius,f(ε)=(ε0-ε)×α,b=4.98×10-16)showed the best predictabilities.The proposed phase-splitting diagram of amine-organic solvent–water systems based on lnγ′of Model 2 was shown in Fig.5.Red groups were those phasesplitting systems after CO2absorption.Blue groups were those systems that remained homogeneous after CO2absorption.A linear boundary of biphasic points and monophasic points could be plotted (Y=0).
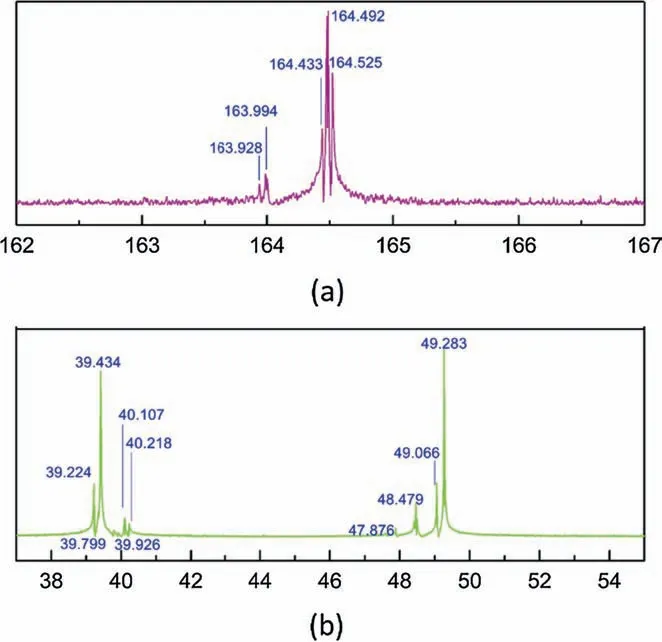
Fig.4.13C NMR spectrum for DETA-1-Propanol-water system.
Among the six outliers,three of them were related to the concentration(No.37,No.48,No.72),and three of them were related to the solvent properties(No.2,No.86,No.91).No.37,No.48 and No.72 outliers appeared under a low concentration,while the same systems could be predicted accurately under other concentrations.With the water concentration increasing,some unex-pected product such as [HCO3-] may appears,then the boundary error may exist for those systems [20].In the MEA-2-Methoxyethanol system (the outlier No.2),the solvent 2-methoxyethanol was the only solvent with both hydroxy and ether functional groups.The PDA-1-propanol system (No.85) and the PDA-1-propanol system (No.90) occurred phase-splitting after absorption,while the PDA-2-propanol system (No.86) and the PDA-2-propanol system (No.91) were still homogeneous after absorption.Model 2 cannot explain the subtle difference between 1-propanol and 2-propanol,while it can be tell in Model 3 considering the interaction between the ions and solvents.The No.85,No.90,No.86 and No.91 were accurately predicted in Model 3.The steric hindrance and the interaction between the ions and solvents impacts phase-splitting behaviors,and those may help to improve the models.Besides those outliers,all the data set were regular.All the tri-amine and bi-amine showed poly-active sites to absorb CO2.The poly-amine systems were prone to occur phase-splitting,while some poly-amine systems remained homogeneous due to the solvent properties.In total,93.5% of the amine-organic solvent–water systems were predicted successfully with Model 2.
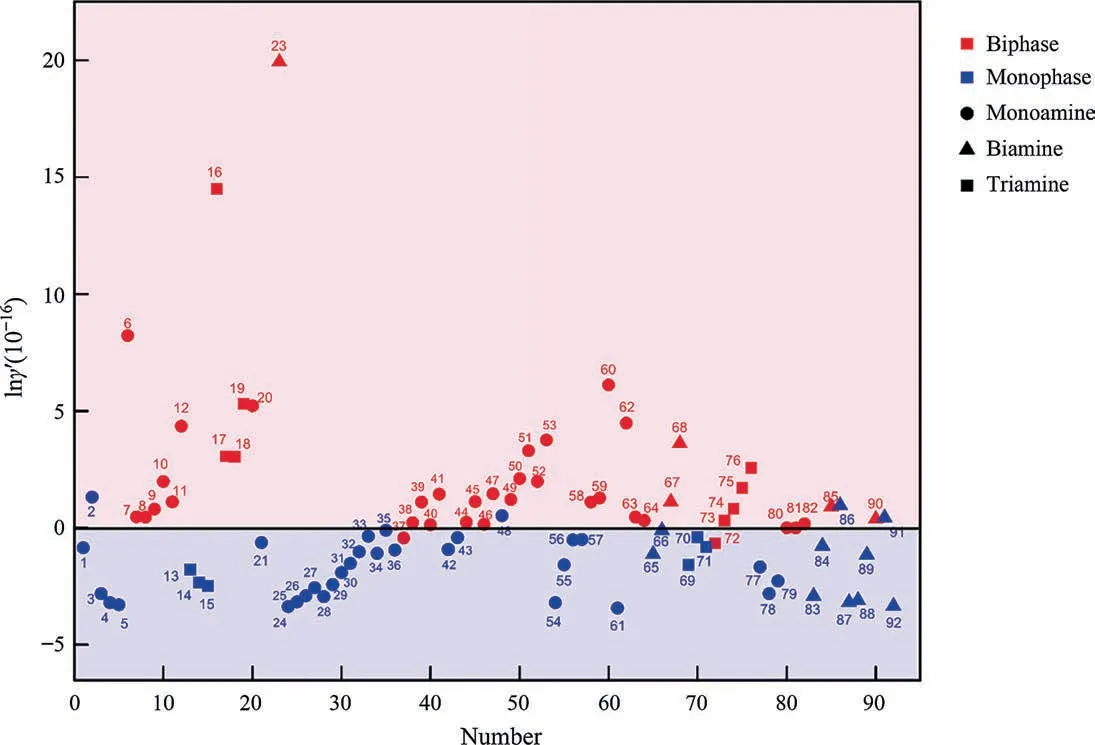
Fig.5.Proposed phase-splitting diagram of amine-organic solvent–water systems based on lnγ′ of Model 2.
The lnγ′expressed as Eq.(13) (b=4.98 × 10-16) could be used for predicting the phase-splitting behaviors of amine-organic solvent -water systems.Those systems with a lnγ′value over 0 were predicted with the phase-splitting behavior,while the systems with a lnγ′value less than 0 placed were predicted to remain homogeneous after CO2absorption.Poly-amine and mono-amine with small molecular volumes would be the good candidates for phase-splitting system.Long-chain alcohol,branched or ring structure,aprotic solvent or solvent with a low static dielectric constant may be suited for a phase-splitting system.This model could apply to the mono-amine system and the poly-amine system with different materials proportions.

5.Conclusions
To predict phase-splitting behaviors for mono-amine or polyamine of the amine-organic solvent–water system with various concentration,a quasi-activity coefficient (lnγ′) model was developed based on Debye &McAulay equation and some DFT descriptors.
Absorption experiments were carried out to confirm the products of a poly-amine system.13C NMR spectrum indicated that bi-carbamate [DETA(COO)2]2-and tri-carbamate [DETA(COO)3]3-may coexist in the system.But the bi-carbamate of primary N dominated.No HCO3-/was detected.
The best lnγ′model was constructed with the ion radius,the descriptors of the volume of organic solvents,the descriptors of the maximum electrostatic potential value of the solvent molecular surface,and the descriptors of the surface/volume value of organic solvents.Considering the ionic geometric configuration and the ion–solvent interactions would help to tell the difference between some isomers such as 1-propanol and 2-propanol to improve model prediction ability.The model could apply to the poly-amines systems with different materials proportions.It would help for further designing phase-splitting absorbent system.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Thanks to the financial support from National Natural Science Foundation of China(21878190)and China Petrochemical Corporation(419033-1).Thanks to the Engineering Experimental Teaching Center,School of Chemical Engineering,Sichuan University for the Nuclear Magnetic Resonance (NMR,JNM-ECZ400S/L1,JEOL Ltd.)support and the compute server support.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2022.02.003.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Green hydrogen:A promising way to the carbon-free society
- Electrochemical CO2 mineralization for red mud treatment driven by hydrogen-cycled membrane electrolysis
- Fabrication of azobenzene-functionalized porous polymers for selective CO2 capture
- Significantly enhanced charge transfer efficiency and surface reaction on NiP2/g-C3N4 heterojunction for photocatalytic hydrogen evolution
- CO2 capture by double metal modified CaO-based sorbents from pyrolysis gases
- Methane hydrate crystal growth on shell substrate