Pyrolyzing soft template-containing poly(ionic liquid) into hierarchical N-doped porous carbon for electroreduction of carbon dioxide
2022-04-27MingdongSunZhengyunBianWeiweiCuiXiaolongZhaoShuDongXuebinKeYuZhouJunWang
Mingdong Sun,Zhengyun Bian,Weiwei Cui,Xiaolong Zhao,Shu Dong,Xuebin Ke,Yu Zhou,*,Jun Wang,*
1 State Key Laboratory of Materials-Oriented Chemical Engineering,College of Chemical Engineering,Nanjing Tech University,Nanjing 210009,China
2 Department of Chemical Engineering,University of Hull,Hull HU6 7RX,United Kingdom
Keywords:CO2RR Poly(ionic liquid)N-doped carbon materials Pore diameter Hierarchical pore
ABSTRACT Heteroatom-doped carbon materials have demonstrated great potential in the electrochemical reduction reaction of CO2(CO2RR)due to their versatile structure and function.However,rational structure control remains one challenge.In this work,we reported a unique carbon precursor of soft template-containing porous poly(ionic liquid) (PIL) that was directly synthesized via free-radical self-polymerization of ionic liquid monomer in a soft template route.Variation of the carbonization temperature in a direct pyrolysis process without any additive yielded a series of carbon materials with facile adjustable textural properties and N species.Significantly,the integration of soft-template in the PIL precursor led to the formation of hierarchical porous carbon material with a higher surface area and larger pore size than that from the template-free precursor.In CO2RR to CO,the champion catalyst gave a Faraday efficiency of 83.0% and a current density of 1.79 mA∙cm-2 at-0.9 V vs.reversible hydrogen electrode(vs.RHE).The abundant graphite N species and hierarchical pore structure,especially the unique hierarchical small-/ultramicropores were revealed to enable better CO2RR performance.
1.Introduction
As one of the main greenhouse gases with a continuous growing concentration in the atmosphere,carbon dioxide (CO2) is indeed a cheap and renewable C1 resource[1,2].Catalytic conversion of CO2into valuable chemicals and fuels is of great significance to alleviate the energy and environmental crisis [3,4].Among various strategies,the electrochemical reduction reaction of CO2(CO2RR)is greatly attractive because of the favorable operation under mild conditions and the advantages in energy storage by using renewable clean energy such as solar and wind [5–7].However,the development of highly effective electrocatalysts remains one challenge due to the sluggishness of inert CO2molecules and the inevitable competing hydrogen evolution reaction (HER) in aqueous media [1].
Various metal-based electrocatalysts,especially noble metals,like Au,Pd,and Ag,have been established for the CO2RR process[8–11],which still suffered from high cost,limited resource,or environmental unfriend [12,13].This situation motivates the development of metal-free electrocatalysts such as non-metallic inorganics [14],biomass [15],polymers [16],carbon materials[13,17–19],etc.Especially,N-dopped porous carbon (NPC) was extensively studied because of the abundant natural resource,large surface area,good conductivity,high stability,and tunable functionality [12,20].Their electro-catalytic performance for CO2-RR associates closely with type,density,dispersion of N species[21],plus porosity and morphology [22,23].Modulation of these structural characters strongly depends on the precursors and the preparation method.For example,N resources were normally externally added to integrate N species into porous carbon via pyrolysis[24].On the other hand,hard/soft template or metal salts activation was used to create abundant hierarchical porosity in NPC for improving mass transfer [25].Despite much progress,it is still challenging to rationally design carbon electrocatalysts with controllable N species and porosity.We note that the facile preparation of NPC via direct pyrolysis of N-containing precursors favors improving the dispersion and stability of N species and fabricating hierarchical porous structures[26].Nonetheless,the qualified carbon precursors to render this issue are rare,so far.
Ionic liquids (ILs),especially the most popular imidazoliumbased ILs,have been employed as the NPC precursors for decades,due to their negligible vapor pressure,high thermal stability,and inherently N-doping structures [27–29].In principle,porous precursors exemplified by porous organic polymers(POPs)are attractive to controllably obtain porous carbon via pyrolysis[30].Porous poly(ionic liquid) (PIL),combining the advantages of ILs and POPs,are emerging as a novel family of carbon precursors with the taskspecific design of cations and anions,facile doping of N species,and possible tuning of pore structures[31].As afore we noticed,N species and porous structure are two key factors for the NPC-based CO2RR process [23],it is thus rational to say that PIL-derived carbon materials are highly desirable for this reaction.Indeed,PILderived carbon materials have been applied in CO2adsorption[32],energy storage [33],and catalysis [34,35].For instance,the N,P dual-doped carbon prepared by pyrolyzing poly[vinyl imidazolium dihydrogen phosphate] was reported as an active metalfree electrocatalyst for oxygen reduction reaction (ORR) [35].For CO2RR to formate,a nanoporous polymer/CNTs membrane was fabricated by coating poly[1-cyanomethyl-3-vinylimidazolium bis(trifluoromethanesulfonyl)imide] on carbon nanotubes followed with an ammonia treatment,the carbonization of which gave a hierarchical porous nitrogen-doped carbon/CNTs composite showing a Faradaic efficiency(FE)of 81%[34].To the best of our knowledge,the application of PIL-derived carbon materials in CO2RR is much limited and to be explored.
In this work,we task-specifically prepared the hierarchically porous NPC material by pyrolyzing the PIL precursor and proved its efficiency for the CO2RR process.The PIL precursor was synthesized from the free radical self-polymerization of the ILs monomer 1-allyl-3-vinylimidazolium chloride in the presence of block copolymer P123 as the soft template,according to our previous work [36].Rather than the pre-removal of the soft template by a solvent,the P123-containing PIL was directly subjected to pyrolysis for obtaining the target NPC.Systematic characterizations suggest that considerable amounts of N species remained at the high pyrolysis temperature beyond 1000°C and the formation of hierarchical pore structure arose from the soft template.For CO2RR,the resultant NPC material exhibited a high FE towards CO (83.0%),and the property-activity relationship was analyzed and discussed in detail.
2.Experimental
2.1.Materials and methods
ILs monomer,1-allyl-3-vinylimidazolium chloride ([AVIM]Cl,≥99%),was purchased from Lanzhou Greenchem ILs,LICP,Chinese Academy of Sciences.Triblock copolymer poly(ethylene glycol)-block-poly (propylene glycol)-block-poly (ethylene glycol) (P123,Mw=5800) was purchased from Sigma-Aldrich.Polyethylene glycol (PEG,Mw=20,000) was bought from Xilong Science Co.Ltd.Ammonium persulfate (APS,>98%) was purchased from Shanghai Lingfeng Chemical Reagent Co.Ltd.Potassium bicarbonate(KHCO3,99.99%) was obtained from Shanghai Macklin Biochemical Co.Ltd.Nafion solution(5.0%(mass))was available from DuPont.All chemical substances were used as received without further purification.
X-ray photoelectron spectroscopy (XPS) was performed on a PHI 5000 Versa Probe X-ray photoelectron spectrometer equipped with Al Kα radiation(1486.6 eV).Thermogravimetric(TG)analysis was carried out on an STA409 instrument under the nitrogen atmosphere at a heating rate of 10 °C∙min-1.X-ray diffraction(XRD) patterns were collected on a Smart Lab diffractometer(Rigaku Corporation) equipped with a 9 kW Cu rotating-anode source.Nitrogen sorption isotherms and pore size distribution curves were measured at -196 °C on a BELSORP-MINI analyzer.Scanning electron microscopy (SEM) images were acquired with a Hitachi S-4800 scanning electron microscope (10 kV).Transmission electron microscopy (TEM) images were collected on a JEOL JEM-2100 apparatus (200 kV).Fourier transform infrared spectra(FTIR)were recorded on an Agilent Cary 660 instrument.Elemental compositions were analyzed on a Vario EL cube CHN elemental analyzer.Raman spectra were recorded on a Horiba HR 800 spectrometer with a Spectra-Physics 2018 Argon/Krypton Ion Laser system (excitation line:514 nm).Quantitative analysis of liquid products after electrolysis of the electrolyte was carried out by nuclear magnetic resonance.
2.2.Materials synthesis
2.2.1.Preparation of PIL precursor
Self-polymerization of [AVIM]Cl was carried out in a modified soft-template pathway following a previous report [36].In a typical synthesis,P123(2 g),PEG(1 g),and H2O(8 g)were added into a 50 ml beaker and stirred at room temperature for 24 h,followed by the addition of [AVIM]Cl (2 g) and additional stirring for 24 h.The polymerization occurred at 40 °C for 10 h and then 50 °C for 14 h after the addition of APS (7 g) as initiator.The formed white solid was isolated by filtration and washed with deionized water,and then vacuum dried at 60 °C to give the PIL precursor,named CAV.For comparison,nonporous counterpart CAV-N was synthesized in the absence of P123 and PEG.
2.2.2.Preparation of PIL-derived carbon materials
PIL-derived carbon materials were prepared by a one-step pyrolysis method by using CAV as the carbon resource.Specifically,CAV (2 g) was placed into a crucible at the center of the atmosphere furnace,the temperature of which was raised to the desired pyrolysis temperatures at a rate of 2 °C∙min-1under N2atmosphere with a flow rate of 200 ml.min-1and maintained for 2 h.The obtained carbon materials were denoted as PAC-T,where T represents the pyrolysis temperature.Thus,carbon materials prepared via pyrolysis of CAV at five different temperatures of 800 °C,900 °C,1000 °C,1100 °C,1200 °C were denoted as PAC-800,PAC-900,PAC-1000,PAC-1100,PAC-1200,respectively.And pyrolysis of CAV-N at 1100 °C gave the carbon materials ofnt-PAC-1100.
2.3.Electrochemical measurements
2.3.1.Preparation of working electrode
Typically,well ground catalyst powder(5 mg)was dispersed in a mixture of Nafion solution(5%(mass),40 μl)and ethanol(400 μl)via ultrasonication for 30 min.Subsequently,the obtained catalyst ink(88 μl)was dropped onto the carbon paper substrate electrode(coating area:1 cm × 1 cm),and then dried at 30 °C to give the working electrode with a catalyst mass loading of 1 mg.cm-2.
2.3.2.Electrochemical reduction of CO2
All the electrochemical experiments were carried out on a CHI 760E electrochemical workstation (Shanghai Chen-Hua,China) by using a gas-tight two-compartment H-cell equipped with a standard three-electrode configuration.The three-electrode systems included a working electrode as prepared above,an Ag/AgCl electrode (filled with 3.5 mol.L-1KCl) as the reference electrode,and a Pt plate (1 cm × 1 cm) as the counter electrode.The former two electrodes were placed in the cathode chamber,while the latter one was placed in the anode chamber.Each compartment was filled with~ 50 ml electrolyte (0.1 mol.L-1KHCO3aqueous solution) and separated by a piece of Nafion 117 proton exchange membrane to avoid the re-oxidation of CO2RR-generated products.The measured potentials were rescaled to the reversible hydrogen electrode(RHE)according to the following equation:E(vs.RHE)=E(vs.Ag/AgCl) +0.2046 V +0.059 V × pH.All the mentioned potentials were referred to the RHE unless otherwise noted.
Before the electrolysis experiment,the cathodic chamber was bubbled with high-purity CO2(99.9999%,40 ml.min-1) for 30 min to remove O2from the electrolyte and saturate the electrolyte with CO2.Electrochemical reduction of CO2was carried out at each applied potential for 30 min under ambient conditions.The current density (J) was normalized to the geometrical area of the working electrode (1 cm2).The gas-phase was analyzed by a gas chromatograph (GC-9860-5CNJ,Nanjing Hope Analytical Equipment Co.,LTD,China) equipped with a flame ionization detector (FID),a thermal conductivity detector (TCD),and a 5A molecular sieve packed column (2 m).Only CO and H2were detected in this study.After the potentiostat electrolysis,800 μl catholyte was mixed with 100 μl D2O and then analyzed by1H nuclear magnetic resonance (1H NMR) spectrometer (Bruker DPX 500 MHz),showing that no liquid product formed.Faraday efficiency (FE) of gaseous products at each applied potential was calculated based on the equation:
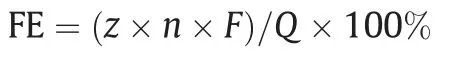
where F is the Faraday constant,96,485 C.mol-1;Q is the total charge acquired from the electrochemical workstation,C;n is the moles of the formed gas product,mol;z is the number of the transferred electron to produce a molecule of the gas product.
Linear sweep voltammetry (LSV) measurements were performed in N2-or CO2-saturated 0.1 mol.L-1KHCO3electrolyte with a scan rate of 5 mV.s-1.The pH of the CO2-saturated 0.1 mol.L-1KHCO3electrolyte was 6.8.Cyclic voltammetry (CV) was performed at the potential range 0 to 0.5 V in an N2-saturated 0.1 mol.L-1KHCO3solution with different scan rates,giving the double-layer capacitance(Cdl)for the determining of electrochemical surface area(ECSA).Cdlvalue is calculated by plotting ΔJ(Ja-Jc)as a function of scan rate,in which Jaand Jcare anode and cathode current densities,respectively.The slope of the line is twice that of Cdl.Electrochemical impedance spectroscopy (EIS) was tested at-0.9 V in the frequency range from 0.01 Hz to 100 kHz with a voltage amplitude of 10 mV.
3.Results and Discussion
3.1.Synthesis and characterization of carbon materials
Fig.1 illustrates the preparation procedure for the PIL CAV and the resultant carbon materials.The precursor CAV was synthesized via the free radical self-polymerization of ionic liquid monomer 1-allyl-3-vinylimidazolium chloride([AVIM]Cl) by using ammonium persulfate(APS)as the initiator and P123 as the soft template.The Fourier transform infrared (FT-IR) spectrum of CAV (Fig.S1)showed characteristic bands of the imidazole group with the disappearance of the C=C bond,evidencing the polymerization of[AVIM]Cl.The thermogravimetric (TG) curve of CAV (Fig.S2) presented a shape weight loss from 200 °C to 500 °C due to the removal of the template with the possible partial decomposition of the polymeric skeleton.The weak weight loss above 500 °C reflects the high thermal stability.Without pre-removing of the soft template,direct pyrolysis of CAV at different temperatures(T=800,900,1000,1100,and 1200) yielded a series of Ndopped carbon materials,termed PAC-T.Various characterization techniques were applied to reveal the structural information of PAC-T.Scanning electron microscopy (SEM) in Fig.2(a) and Fig.S3 demonstrated that these carbon materials are composed of highly fused primary particles,which are sized in hundreds of nanometers and loosely packed to form secondary aggregates at the micrometer level.The morphologies of the PAC-T series are similar to each other and resemble that of parent CAV.The internal lattice spacing of the (0 0 2) plane of graphitic carbon is 0.34 nm and would slightly expand after the heteroatom doping.Therefore,the crystal plane in Fig.2(c),with the lattice spacing of 0.38 nm is assigned to (0 0 2) plane,attributable to N doping.[37].SEM elemental mapping images manifested a homogeneous C,N,and O distribution over the representative PAC-1100 (Fig.2(d)–(g)).It can be seen from the XRD patterns (Fig.3(a)) that all the PAC-T samples exhibited two broad diffraction peaks at 2θ ≈26° and 44°,belonging to the(0 0 2)and(1 0 0)planes for graphitized carbon,respectively [38,39].As the temperature increased,the peaks gradually became weaker,indicating the increased disorder degree of the graphite structure.Raman spectra(Fig.3(b))showed the two distinctive peaks 1340(D band)and 1590 cm-1(G band)relative to defective/disordered carbon and crystalline graphitic sp2-carbon,respectively [40–42].The ID/IGvalues were calculated from the intensity ratio of D to G bands to quantitatively reflect the disorder degree of carbon materials.The slight increase in ID/IGvalues from 0.96 to 1.16 along with the carbonization temperatures from 800 °C to 1200 °C again suggests the formation of more defects at a higher temperature.
The textural properties including the specific surface area,porevolume,and pore size were characterized by the N2sorption experiment.As shown in Fig.4(a),N2uptakes jump at relatively low pressure occurred in all the nitrogen sorption isotherms,reflecting the formation of a large number of micropores.Relatively weak uptakes were observed over the samples prepared at the low pyrolysis temperatures of 800 °C and 900 °C,and the typical type I isotherms hint the classical full microporous structure which is confirmed by the single pore-distribution in Fig.4(b).The hightemperature pyrolyzed samples (PAC-1000,PAC-1100,and PAC-1200),showed remarkably higher uptake values with the secondary enhancement at P/P0>0.4,suggesting hierarchical pore structures [43].Fig.4(b) shows that PAC-1000 and PAC-1200 remained the similar micropore diameter to PAC-800 and PAC-900,and added the other most probable mesopores,demonstrating the hierarchically micro-/meso-pore structure.Interestingly,PAC-1100 gave the smaller micropore at 0.5–0.6 nm also called ultramicropore,with the appearance of the other large micropore at 1–2 nm,demonstrating an unusual bimicro-sized hierarchical pore structure.Further,among all the samples,PAC-1100 owned the maximum surface area(2257 m2.g-1)and pore volume(1.57 cm3-.g-1),associating well to its plenty of ultra-micropores and hierarchical pore structure (Table S1).During the carbonization of PIL,two following opposite influencing factors dominate the pore formation and variation.The decomposition of the organic skeleton and the removal of formed gas (CxHy,NOx,and SOx,etc.) initiated the pore formation[44].As the temperature increased,the shrinkage and decomposition of the skeleton occurred simultaneously,and at high temperatures,more remarkable collapse relative to shrinkage tended to result in the generation of larger pores[44,45].In this work,at relatively low temperatures,the decomposition effect is greater than the shrinkage,leading to the formation of main micropores.Carbonization at moderate temperatures(e.g.1100 °C) reached a balance of the above two factors,affording the hierarchical porous structure,in this case,the removal of formed gases serving as pore formation agents may create ultramicropores.The containing of a soft template in our PIL precursor made this pyrolysis carbonization unique,which may account for the creation of the unusual hierarchical small-/ultra-micropores.The excessive-high temperatures (e.g.1200 °C) caused severe decomposition and then collapse of the organic skeleton,giving rise to reducing of the ultra-micropores and increase of mesopores.Further,among all the samples,PAC-1100 owned the maximum surface area (2257 m2.g-1) and pore volume (1.57 cm3.g-1),associating well to its plenty of ultra-micropores and hierarchical pore structure(Table S1).Hierarchical micro-mesoporous carbon materials have been reported by using various precursors[44,46].Compared with these materials,the optimal sample PAC-1100 had hierarchical small-/ultra-micropores with a larger surface area,which are advantageous in the mass transfer for CO2RR to CO.Besides,carbonization of PIL have been employed to produce porous carbon materials,but the formation of such type pores and the application in CO2RR was not reported.Herein,we demonstrated that modulation of the temperature and structure of precursors enabled the formation of specific carbon material of PAC-1100,resulting in superior activity in the CO2RR to CO (details are seen in the section of Electrochemical CO2reduction below).

Fig.1.Synthetic route of porous poly(ionic liquid) CAV and the corresponding derived carbon materials.

Fig.2.(a) SEM image,(b),(c) TEM images,and (d)–(g) SEM elemental (C,N,and O) imaging of PAC-1100.

Fig.3.(a) XRD patterns and (b) Raman spectra of PAC-T series.

Fig.4.(a) N2 sorption isotherms (b) corresponding pore size distribution curves of PAC-T series.
The C,H,N elemental compositions of CAV and PAC-T series were measured (Table S2),showing that the elemental composition of CAV was C 38.7%,N 8.1%,and H 6.2% (entry 1).After pyrolysis,the C content of PAC-T samples increased while the N contents decreased,attributable to the preferential decomposition of the heteroatomic N-containing functional groups at high temperatures.The surface chemical composition and electronic state were analyzed by X-ray photoelectron spectroscopy (XPS) (Fig.5(a) and(b)).The survey scan XPS spectra(Fig.5(a))displayed the signals of C,N,and O elements for all the samples.The high-resolution N1s XPS spectra (Fig.5(b)) were deconvoluted into the four peaks at 398.5,399.9,401.3,and 404.0 eV assigned to pyridinic N (Pyri-N),pyrrolic N (Pyro-N),graphitic N (Grap-N),and oxidized N(Oxid-N),respectively [47,48].The high-content N species are Pyri-N and Pyro-N at the low pyrolysis temperatures of 800 °C and 900 °C (Table S3).Consequently,the degree of graphitization is insufficient in that case,and N atoms are distributed at the edges and fractures of the carbon layer to yield a large amount of Pyri-N and Pyro-N.Higher temperatures lead to the partial removal of N atoms and promote the re-embedding of the preserved N atoms into the carbon layer [41,49,50].As a result,when the pyrolysis was conducted at 1000 °C and above,Grap-N became dominating the N species,which was beyond 62% into a more stable Grap-N.PAC-1100 exhibited the highest Grap-N content of 71.0%,but the extremely high pyrolysis temperature 1200 °C caused a jump of Oxid-N with a reversely lowered Grap-N down to 62.8%.The results indicate that 1100 °C is an optimal pyrolysis temperature to obtain a sufficiently graphitized carbon material from the CAV precursor.
For comparison,self-polymerization of ILs monomer [AVIM]Cl in the absence of template (P123) was carried out under the otherwise same conditions as those of CAV,giving a nonporous PIL material.Carbonization of this PIL at 1100 °C reached a control samplentPAC-1100.Compared with PAC-1100,ntPAC-1100 exhibited a lower surface area (1680 m2.g-1) and pore volume(0.58 cm3.g-1) (Table S1).Its nitrogen sorption isotherm was the typical type I for microporous materials (Fig.S4).Only slight increasing uptake at high relative pressure above 0.5 was observed,suggesting negligible mesopores.The porosity ofnt-PAC-1100 was in contrast to the hierarchically porous structure of the soft template-derived PAC-1100.As demonstrated previously,removing the soft template from the as-synthesized PIL by using a solvent is difficult[36],which,however,is unnecessary in preparing carbon materials by the present pyrolysis.The above comparison indicates that the template-containing PIL material is favorable for the formation of hierarchically porous carbon,mostly due to the lower decomposing temperature of the polymeric soft template over the PIL backbone.Elemental and XPS analysis ofntPAC-1100 demonstrated that the total N content and surface chemical status of the four N species are comparable to those of PAC-1100,implying that the modulation of N species should have not been affected by the inclusion of the soft template (Fig.S5).Thus,the soft template-containing PIL acts as an interesting precursor that provides an opportunity to control the hierarchical pore formation while featuring the inherently created surface chemical state of N species.
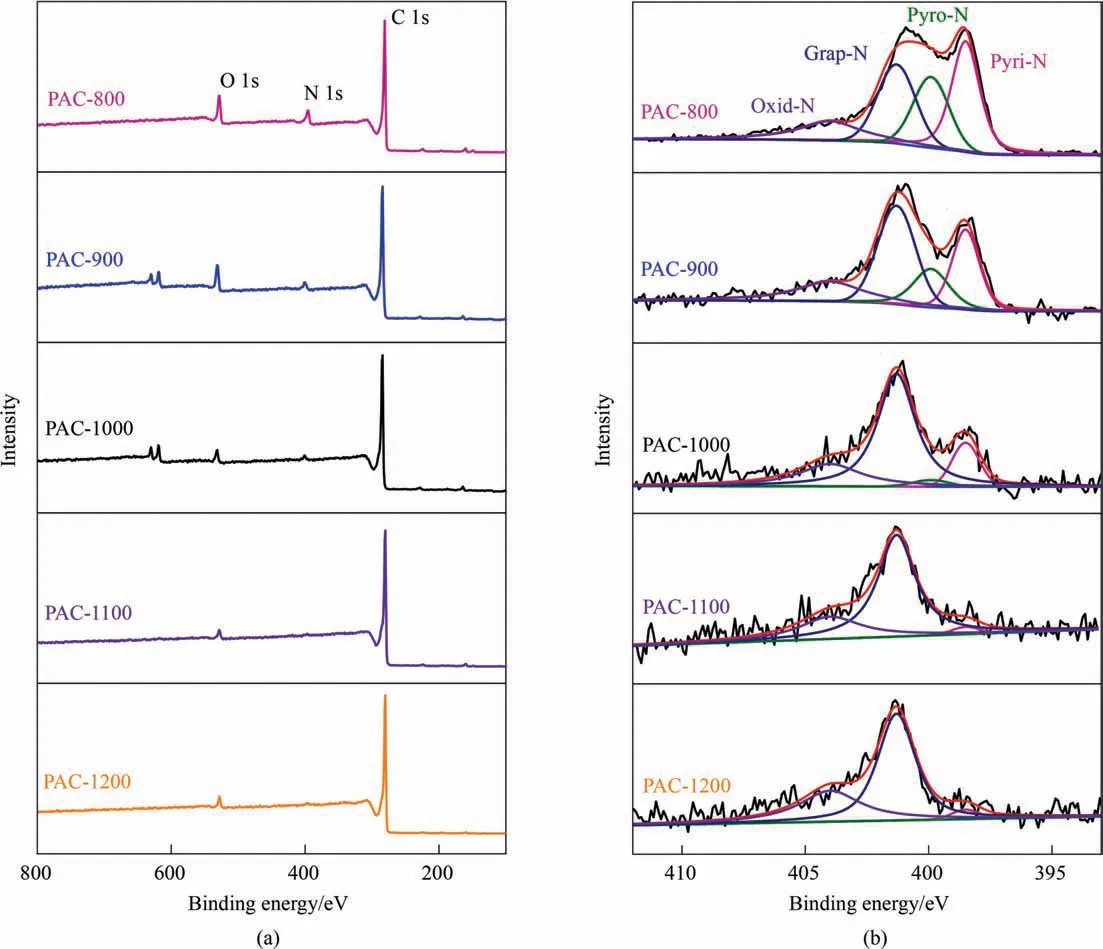
Fig.5.(a) Survey scan XPS spectra,and (b) high-resolution N 1s XPS spectra of PAC-T series.
3.2.Electrochemical CO2 reduction
The CO2RR on the PAC-T series was assessed in an H-type cell by using a three-electrode system.In linear sweep voltammetry(LSV)curves of PAC-1100 with N2saturated 0.1 mol.L-1KHCO3electrolyte(Fig.6(a)),the onset potential was-0.9 V,implying a strong ability to inhibit hydrogen evolution reduction (HER).By contrast with CO2saturated electrolyte,the onset potential positively shifted to the lower region,and larger current density was observed,indicating the taking place of CO2reduction.
The CO2RR on the PAC-T series was further measured in the cathode potential range of -0.5 to -1.1 V.CO and H2turned out to be the main reduction products with the total Faradaic efficiencies(FEs)of around 100%and no liquid product was detected by1H NMR(Fig.S6).Fig.6(b)and Fig.S7 respectively illustrate FE(CO)(FE of CO) and FE(H2) (FE of H2) over PAC-T series for CO2RR at the applied potentials.PAC-1100 showed the FE(CO) of 4.4% at the low potential of-0.5 V,which gradually increased with increasing the potential,reaching the maximum of 83.0% at -0.9 V.Further increasing the potential caused the declining of FE(CO)but dramatically enhancing of FE(H2),indicating that HER dominated the reduction at the high potential.The phenomena allow a facile alteration of the CO/H2ratio to selective produce CO or syngas via controlling the applied potential.A similar trend was observed over other PAC-T samples.PAC-1100 exhibited the highest FE(CO)at all the employed potentials.Compared with PAC-1100 exhibiting the maximum FE(CO) of 83.0% at the potential of–0.9 V,PAC-800,PAC-900,PAC-1000,and PAC-1200 showed the FE(CO)of 3.9%,12.2%,59.5%,and 71.1%,respectively.The FE(CO)gradually increased along with the pyrolysis temperature,reaching the maximum value at 1100 °C and then decreasing at the higher temperature.Fig.6(c) shows the CO partial current density JCO,(normalized by the geometrical surface area) on the PAC-T series against the applied potential.PAC-1100 displayed the largest JCOin the entire applied potential range,reflecting the fastest reaction kinetics in CO2RR.The JCOon PAC-800 was nearly zero,in line with its sluggish catalytic performance in CO2RR at the low pyrolysis temperature.Along with the increase of pyrolysis temperature,PAC-1100 afforded the JCOof 1.79 mA.cm-2at the potential of–0.9 V,again reflecting the efficiency in tuning the CO2RR activity by manipulating the pyrolysis temperature.Compared with typical literature published recently [23,42,51],most of those carbon materials exhibited FE(CO) values of 80%–90% and current densities of 1–3 mA.cm-2.One can see that PAC-1100 afforded comparable efficiency to those carbon materials,suggesting that it was a potential robust carbon catalyst for CO2RR to CO(Table S4).A longterm CO2RR performance on PAC-1100 was carried out at the constant cathode potential of -0.9 V vs.RHE (Fig.6(d)).Only a slight decay of FE(CO) and JCOwas observed in the initial stage,after which they became quite stable up to 20 h.
For comparison,CO2RR overntPAC-1100 was evaluated in potentials from -0.6 to -1.0 V .The maximum FE(CO) of 61.2%with the JCO0.76 mA.cm-2at the potential of -0.9 V was much lower than those on PAC-1100.The comparison visualizes the advantage of the utilization of porous PIL as the carbon precursor for CO2RR (Fig.S7).

Fig.6.Catalytic CO2RR performance.(a) LSV curves of PAC-1100 measured in N2-and CO2-saturated electrolyte (0.1 mol.L-1 KHCO3 solution) with a scan rate of 5 mV.s-1.Faraday efficiency of (b) CO and (c) CO partial current densities of PAC-T series.(d) Stability tests of PAC-1100 at–1.5 V vs.RHE in CO2 saturated electrolyte (0.1 mol.L-1 KHCO3 solution).
To ascertain the CO production from the CO2RR process rather than the reduction of KHCO3or the decomposition of catalysts,potentiostat electrolysis on PAC-1100 was performed in N2saturated 0.1 mol.L-1KHCO3electrolyte at -0.9 V (Fig.S8).When the CO2saturated electrolyte was replaced by N2saturated one,FE(CO)dropped from 83.0%to 1.1%with the main product of H2,confirming the origination of CO from CO2reduction.Further,H2formation with negligible FE(CO) was found for the electrolysis on carbon paper in CO2saturated 0.1 mol.L-1KHCO3electrolyte,emphasizing the key role of electrocatalyst PAC-1100 (Fig.S9).
3.3.Understanding of electrocatalysis performance
For CO2RR,neat carbon has been reported to be inactive,while the content and type of N dopant play a crucial role in the action[51].There mainly exist Pyri-N,Pyro-N,and Grap-N species that are potential active sites for N-dopped carbon in CO2RR.All of them may activate CO2molecules via strong affinity.In general,the N content plays a minor role,while the proportion of different N species contributes more significantly [52].However,it remains a big challenge to identify the role of these N species by providing enough direct evidence.Previous studies indicated that Pyri-N species were more active,whereas also some investigations pointed out that Grap-N species were able to provide a lower CO2activation barrier [22,23,53].Besides,it was reported that Pyri-N or Grap-N alone or in a higher proportion can improve electron transfer and optimize the interface microchemical environment,but the existence of the two N species in a considerable amount would greatly weaken this effect [54].In this work,the total N content decreased with the elevating of pyrolysis temperature,but still provided a considerable amount of active sites,while the proportion of different N species varied.The Grap-N content of PAC-1100 is indeed slightly lower than that of PAC-1000,but the former exhibited better performance than the latter,in line with the crucial role of high proportioned Grap-N species.For PAC-T of this work,the variation of the maximum FE(CO) resembles that of Grap-N proportion,whereas no such regulation is required for Pyri-N,Pyro-N,and Oxid-N(Fig.S10a).Based on this phenomenon and previous studies,we speculated that Grap-N species served as the major active sites to activate CO2molecules.On the one hand,Grap-N species plays a positive role in improving the conductivity of carbon materials.On the other hand,Grap-N species favor providing a strong affinity towards the key intermediate*CO2such that the energy barrier is lowered and the activity is enhanced[53,55].Further,the exposure and accessibility of the N active sites is another key factor influencing the CO2RR performance[51].The variation of the maximum FE(CO)on PAC-T series is similar to that of the specific surface area (Fig.S10b),where PAC-1100 with the highest specific surface area possessed the best FE(CO).It is interesting to note the unusual small-/ultra-microporous hierarchical structure for PAC-1100,whereas its analogs owned more popular micro-/meso-porous hierarchical structures.The ultra-micropore size of PAC-1100 was 0.5–0.6 nm,slightly larger than the dynamic diameter of the CO2molecule.In this case,the nano-confinement effect is remarkable,increasing the collision probability of CO2molecule with pore wall accommodating the active sites.This ultra-micropore confinement effect is unique for PAC-1100,and plays an exclusively positive role in improving the CO2RR reactivity.This relationship is confirmed by the comparison of PAC-1100 withntPAC-1100;both of them owned more or less similar surface chemical states but the latter with the lower specific surface area was less active.

Fig.7.(a)Double-layer capacitance,(b)Tafel plots for CO formation,(c)EIS at–1.5 V vs.RHE,and(d)CO2 adsorption isotherms of PAC-900,PAC-1000,PAC-1100,PAC-1200,and ntPAC-1100 at 298 K.
In order to more comprehensively interpret the importance of the exposure and accessibility of active sites,the electrochemical surface area (ECSA) was calculated from the double-layer capacitance (Cdl) by collecting the cyclic voltammograms (CV) curves at varied scanning rate (Fig.S11).For PAC-900,the ECSA value was a high as 90 mF.cm-2,attributable to the high total N content though with a low specific surface area.With increasing pyrolysis temperature,the higher ECSA of 96 mF cm-2was observed for PAC-1000 and PAC-1100,a compromised result of the increase of specific surface area and the decrease of N content.Further raising the pyrolysis temperature to 1200 °C caused a drop ECSA down to 82 mF.cm-2due to reducing the specific surface area and N content.Besides,ntPAC-1100 only showed the ECSA 69 mF.cm-2mainly because of its very small specific surface area.It is thus rational that the best CO2RR activity of PAC-1100 should associate with its most abundant Grap-N species,as well as the highest specific and electrochemical surface areas that suggest good exposure and ready accessibility of these active sites (Fig.7(a)).
The electrocatalytic kinetics of various catalysts in the CO2RR process were evaluated by Tafel slopes,which were calculated by linearly plotting the overpotential vs.logjCO(logarithm of jCO) in the initial low overpotential range (Fig.7(b)).PAC-1100 exhibited a Tafel slope of 114 mV.dec-1,very close to the theoretical value of 118 mV.dec-1with the rate-determining step of the initial transfer of one electron to CO2to form surface adsorbed*CO2.-intermediate [56].The Tafel slope of PAC-1100 was higher than those of other PAC-T members.These data suggest the fastest catalytic kinetics of PAC-1100.Moreover,the electrochemical impedance spectroscopy (EIS) of these samples was measured at the overpotential of-0.9 V(Fig.7(c)).In a Nyquist diagram,the first semicircle diameter represents the ohmic resistance for internal conductivity,and the second one represents the interface charge transfer resistance,the charge transfer resistance between the catalyst and reactant/intermediate,reflecting the diffusion rate,adsorption of reactants,and desorption of products.As shown in Fig.7(c),PAC-900 had a large radius of the first semicircle,revealing the poor inherent conductivity due to the inferior graphitization.The small radius of the first semicircles was observable for other samples,index of favorable conductivity.The sequence of the second semicircle is identical to their CO2RR activity,with PAC-1100 offering the smallest radius,indicative of the lowest interface charge transfer resistance.This EIS observation on PAC-1100 is also advantageous for promoting its reaction kinetic,in good agreement with the above Tafel slope result [42].
As revealed above,the fast kinetics and high activity of PAC-1100 could be tentatively ascribed to plenty of Grap-N sites with good exposure and readily accessibility.Thus,it seems that the champion catalyst should have the best CO2adsorption capability[57].Nonetheless,PAC-1100 did not show the highest CO2uptake among PAC-T series measured by the adsorption isotherms at 298 K (Fig.7(d)).When fitted with a double-site Langmuir (DL)model,two types of sites were demonstrated over PAC-T for capturing CO2(Fig.S12):weak and strong adsorption sites.With the elevation of pyrolysis temperature,the saturated CO2uptake on the abundant weak adsorption sites (qc) gradually increased,accompanied by the decrease of the equilibrium constant kc(affinity coefficient of the weak site).Meanwhile,a reverse trend was found for the strong adsorption sites(qiand ki).This is understandable because the high-temperature pyrolysis reduces the content of N species for strong adsorption sites but greatly increases the specific surface area bearing larger amounts of weak sorption sites.Owing to the combined effects of the two sites,all of the PAC-T samples exhibited considerably high CO2uptakes[58].Particularly,ntPAC-1100 showed similar CO2adsorption isotherms and DL parameters to those of PAC-1100,though the former possessed a much lower specific surface area.Previous reports have shown that,in addition to the specific surface area,hierarchically porous structures for N-doped carbon materials were more crucial for CO2RR,in which large pores improved mass transfer and small pores benefited CO2enrichment nearby the N active sites [59].
4.Conclusions
A series of N-doped porous carbon materials were prepared by pyrolysis of the soft template-containing PIL precursor,which was synthesized by free radical self-polymerization of an ionic liquid monomer in the presence of the soft-template P123.By adjusting pyrolysis temperatures,porosity and N species were modulated for the carbon materials;the pyrolysis at 1100 °C afforded the champion sample PAC-1100 with the maximum specific surface area and Grap-N proportion,as well as the unique hierarchically small-/ultra-microporous structure.PAC-1100 was effective in the CO2RR into CO,exhibiting considerably high Faraday efficiency and current density plus well duration.The identification of the structure-performance relationship suggests that the Grap-N species served as the major sites to activate CO2molecules.The abundant porosity and accessible Grap-N species were observed to promote the activity by facilitating the kinetics and providing a large electrochemical surface area.Specifically,the unique hierarchically small-/ultra-micropore was crucial for the high activity:the small-micropore promoted mass transfer while the nanoconfinement effect of ultra-micropore of 0.5–0.6 nm enhanced the collision probability of CO2with active sites,thereby improving the reactivity of CO2.This work fabricates the first example of soft template-containing PIL-derived hierarchically N-doped porous carbon material for CO2RR towards CO and highlights the importance of the internal porosity of the precursor in controlling the structure and function of the carbon materials.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We gratefully acknowledge support from the National Natural Science Foundation of China (Nos.22072065,U1662107,and 21476109),Six talent peaks project in Jiangsu Province (JNHB-035),State Key Laboratory of Materials-Oriented Chemical Engineering (KL17-04),Jiangsu Provincial Science Foundation for Youths (SBK2020044703),the Project of Priority Academic Program Development of Jiangsu Higher Education Institutions(PAPD),High-Performance Computing Center of Nanjing Tech University for supporting the computational resources and the public service platform for the industrialization of innovative achievements in the field of industrial catalysis.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2021.07.020.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Green hydrogen:A promising way to the carbon-free society
- Electrochemical CO2 mineralization for red mud treatment driven by hydrogen-cycled membrane electrolysis
- Fabrication of azobenzene-functionalized porous polymers for selective CO2 capture
- Significantly enhanced charge transfer efficiency and surface reaction on NiP2/g-C3N4 heterojunction for photocatalytic hydrogen evolution
- CO2 capture by double metal modified CaO-based sorbents from pyrolysis gases
- Methane hydrate crystal growth on shell substrate