可交联嘌呤类脱氧核苷类似物的合成
2022-04-26许永娣马星光孙亚伟
许永娣, 马星光, 孙亚伟
[中国石油大学(华东) 化学化工学院,山东 青岛 266580]
核酸药物是一类能够直接作用于致病靶基因的药物,可在分子水平上调控致病基因的表达,目前已经成为临床医药学的重要组成部分[1-2]。核酸药物的发展对非天然核苷提出了更高的需求,科研工作者们设计并合成了大量不同结构和功能的非天然核苷和非天然寡聚核苷酸来满足研发基因药物的需求。
非天然寡聚核苷酸的结构修饰策略大致分为3类,一是对核糖或脱氧核糖单元进行化学修饰或结构改造,比如在核糖2′-位引入不同构象的卤素原子、醚基、胺基或者烷基单元[3-5],以改善其形成高级结构的化学和生物学稳定性;二是对核苷酸中磷酸二酯进行化学结构改造,比如将其中的磷氧双键替换为磷硫双键,可有效改善核酸药物的抗酶降解能力[6-7];三是对核苷酸上的碱基进行官能团修饰或结构改造,来调节核酸高级结构形成的热力学和动力学,以及改善其抗酶切能力[8-9]。
上世纪90年代,Shigeki Sasaki课题组[10-11]提出,在天然核苷的碱基上引入乙烯基团,可使其与临近核苷上碱基中的胺基发生Michael反应,实现碱基之间的交联。他们将可交联核苷修饰到寡聚核苷酸结构中,通过DNA的链间交联实现基因的定点诱变,从而进行基因表达的调控。该类型的可交联核苷在分子生物学,基因药物以及DNA材料等领域表现出很大的应用价值[12-13]。研究中发现,乙烯基团的高化学活性使得其无法直接用于DNA的化学合成和修饰,因此需要对其进行适当的保护。目前对乙烯基的保护策略是使用甲硫基作为保护单元,该基团在DNA固相合成中的氧化步骤中会被氧化为亚砜基团,而亚砜基团在DNA氨解时会发生β消除,暴露出乙烯基(Chart 1)。

Chart 1
近年来,随着研究人员对可交联类核苷及相关衍生物研究的不断深入,其市场需求也增长,因此对原有的合成路线进行工艺改进和优化具有十分重要的现实意义。目前可交联类核苷的合成方法是以脱氧鸟苷(dG)为原料,经过5步单元反应来实现(Scheme 1)。该方法工艺路线成熟,但是原料成本较高,工艺过程较为繁琐。为了简化纯化步骤,优化合成工艺,本论文以商业化的1-氯-2-脱氧-3,5-二-O-对甲苯甲酰基-D-呋喃核糖为原料,将五步合成缩短为四步,并使用结晶来对中间体进行纯化,以较好的收率实现了目标产物的合成,为大规模生产该类化合物提供了新的思路。
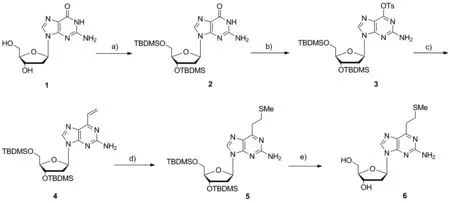
Scheme 1
1 实验部分
1.1 仪器与试剂
RE52CS型真空旋转蒸发器(上海亚荣生化仪器厂);DF-101S型集热式恒温磁力搅拌器(巩义市予华仪器有限责任公司);Bruker AMX-400 MHz型核磁共振谱仪(DMSO-d6为溶剂,TMS为内标);6510 Q-TOF型质谱仪;XT-4型显微熔点仪。
1-氯-2-脱氧-3,5-二-O-对甲基苯甲酰基-D-核糖、2-氨基-6-氯-嘌呤、二(三甲基硅基)氨基钠,氯化锂、四(三苯基膦)钯、三丁基锡乙烯、氯化铵、20%甲硫醇钠溶液、无水乙腈,分析纯,上海泰坦科技股份有限公司;二氯甲烷、1,4-二氧六环、甲醇、氨水、乙酸乙酯、无水硫酸钠,分析纯,国药集团化学试剂北京有限公司。
1.2 合成
(1) 化合物8的合成
将30.7 mL(27.7 mmol)的二(三甲基硅基)氨基钠分散在100 mL乙腈中,搅拌下于55 ℃加入16.9 g(100 mmol) 2-氨基-6-氯-嘌呤,反应2 h。将44.6 g(120 mmol) 1-氯-2-脱氧-3,5-二-O-对甲基苯甲酰基-D-核糖分散在200 mL乙腈中,一次性加入到反应体系中,搅拌下于55 ℃反应8 h(TLC检测)。向反应液中加入10 mL水和5 mL饱和氯化铵溶液淬灭反应。旋蒸除去乙腈,残余物加入400 mL乙酸乙酯和200 mL水,分液,有机相浓缩,残余物经硅胶柱层析[V(石油醚):V(乙酸乙酯)=1∶1]纯化。
化合物8: 白色固体,收率65.5%, m.p.55 ℃;1H NMRδ: 2.33(s, CH3, 3H), 2.36(s, CH3, 3H), 2.69~2.73(m, ArH, 1H), 3.17~3.22(m, ArH, 1H), 4.49~4.53(m, CH2, 2H), 4.58~4.62(m, ArH, 1H), 5.71~5.72(t, ArH, 1H), 6.36~6.38(q, ArH, 1H), 6.99(s, CH2, 2H), 7.25~7.27(d, ArH, 2H), 7.32~7.33(d, ArH, 2H), 7.79~7.81(d, ArH, 2H), 7.89~7.90(d, ArH, 2H), 8.32(s, ArH, 1H);13C NMRδ: 21.71, 35.96, 64.60, 75.56, 82.20, 83.96, 124.23, 127.03~127.09, 129.81~129.98, 141.66, 144.36~144.64, 150.25, 154.21, 160.35, 165.73, 166.00; ESI-MSm/z: Calcd for C26H24ClN5O5{[M+H]+}522.1466, found 522.1539。
(2) 化合物9的合成
称取1.54 g化合物8,780 mg(0.7 mmol)四(三苯基膦)钯和270 mg(6.4 mmol)氯化锂分散在20 mL 1, 4-二氧六环中,搅拌20 min后加入7.8 mL(26.7 mmol)三丁基锡乙烯,体系升温回流15 h。反应结束后,向体系中加入50 mL乙酸乙酯和20 mL氨水,有机相用饱和食盐水洗涤后,加入无水硫酸钠干燥,粗产品用柱层析分离纯化(V(二氯甲烷)/V(乙酸乙酯)=3/1)。
化合物9: 黄色固体,收率80%, m.p.43~44 ℃;1H NMRδ: 2.33(s, CH3, 3H), 2.37(s, CH3, 3H), 2.46~2.47(t, ArH, 1H), 2.68~2.72(m, ArH, 1H), 3.19~3.24(m, ArH, 1H), 5.71~5.72(q, ArH, 1H), 5.80~5.83(q, ArH, 1H), 6.52(s, CH2, 2H), 6.77~6.81(q, ArH, 1H), 6.90~6.95(q, ArH, 1H), 7.26~7.28(d, CH2, 2H), 7.33~7.34(d, CH2, 2H), 7.51~7.53(q, ArH, 1H), 7.57~7.64(td, ArH, H), 7.81~7.82(d, ArH, 2H), 7.90~7.91(d, ArH, 2H), 8.27(s, ArH, 1H);13C NMRδ: 21.76, 35.88, 64.67, 75.68, 82.06, 83.56, 125.10, 125.74, 127.07, 127.12, 129.84, 130.00, 133.15, 141.02, 144.36, 144.64, 153.79, 154.17, 160.75, 165.74, 166.03; ESI-MSm/z: Calcd for C28H27N5O5{[M+H]+}514.2012, found 514.2085。
(3) 化合物10的合成
称取620 mg化合物9分散在20 mL无水乙腈中,然后加入700 μL 20%的甲硫醇钠水溶液,30 min后TLC检测反应完全。旋蒸除去乙腈溶剂,然后向体系中加入50 mL乙酸乙酯和50 mL水,有机相用50 mL饱和食盐水洗涤之后用无水硫酸钠干燥,粗产品以待下一步反应。
化合物10: 淡黄色固体,收率89%, m.p.48~49 ℃;1H NMRδ: 2.05(s, CH3, 3H), 2.33(s, CH3, 3H), 2.36(s, CH3, 3H), 2.66~2.70(m, ArH, 1H), 2.88~2.90(t, CH2, 2H), 3.07~3.10(t, CH2, 2H), 3.18~3.23(m, ArH, 1H), 4.49~4.52(t, CH2, 2H), 4.59~4.62(m, ArH, 1H), 5.70~5.72(m, ArH, 1H), 7.27~7.28(t, ArH, 2H), 7.33~7.34(d, ArH, 2H), 7.82~7.83(d, ArH, 2H), 7.90~7.91(d, ArH, 2H), 8.19(s, ArH, 1H);13C NMRδ: 15.08, 21.72, 21.76, 31.71, 32.81, 35.83, 64.68, 75.70, 82.02, 83.56, 126.38, 127.07~127.13, 129.85~129.99, 140.07, 144.36, 144.63, 152.90, 160.76, 161.13, 165.74, 166.03; ESI-MSm/z: Calcd for C29H31N5O5S{[M+H]+}562.2045, found 562.2119。
(4) 化合物6的合成
将上一步粗产品浓缩溶于10 mL二氯甲烷和4 mL饱和氨的甲醇溶液中,将体系置于耐压反应管内80 ℃搅拌反应,反应时间8 h,得到的粗产品通过乙酸乙酯结晶可得到纯品。
化合物6: 白色固体,收率83%, m.p.79~80 ℃;1H NMRδ: 2.05(s, CH3, 3H), 2.17~2.21(m, ArH, 1H), 2.46~2.47(t, ArH, 1H), 2.57~2.62(m, ArH, 1H), 2.88~2.91(t, CH2, 2H), 3.07~3.09(t, CH2, 2H), 3.45~3.49(m, ArH, 1H), 3.52~3.55(m, ArH, 1H), 3.78~3.80(m, ArH, 1H), 4.32~4.34(m, ArH, 1H), 4.94~4.96(m, ArH, 1H), 5.26(d, ArH, 1H), 6.19~6.22(q, ArH, 1H), 6.44(s, CH2, 2H), 8.18(s, ArH, 1H);13C NMRδ: 15.07, 31.74, 32.77, 62.25, 71.31, 83.12, 88.15, 126.29, 140.06, 152.84, 160.66, 160.86; ESI-MSm/z: Calcd for C13H19N5O3S{[M+H]+}326.12086, found 326.1281。
2 结果与讨论
2.1 碱种类以及用量的优化
在文献报导的可交联核苷类似物的合成方法中,Shigeki Sasaki课题组选择脱氧鸟苷(dG)作为起始原料。脱氧鸟苷价格较高,而且其在多种有机溶剂中溶解性均很差,操作较为困难。本文选择了商业化的1-氯-2-脱氧-3,5-二-O-对甲苯甲酰基-D-呋喃核糖作为起始原料,通过SN2反应将鸟嘌呤以C—N糖苷键连接到脱氧戊糖环上,构建脱氧鸟苷类似物(Scheme 2)。本文考察了不同类型的碱在反应中的作用[14](表1),以及反应时间和反应温度对于目标产物收率的影响,最终选择二(三甲基硅基)氨基钠作为碱,在55 ℃下反应8 h能以较高的收率得到目标产物。

表1 有机碱种类对化合物8产率的影响
2.2 有机锡试剂的用量的优化
在鸟嘌呤可交联核苷类似物的合成过程中,需要使用三丁基乙烯基锡作为乙烯基试剂,通过Stille反应将嘌呤6-位上的氯转化为乙烯基[15]。文献报道中有机锡试剂使用量为原料物质的量的6倍,有机锡试剂价格昂贵,且化学毒性较大,因此我们希望减少其在合成中的使用量。本文考察了反应中有机锡的使用量,表2中的实验结果证实,将有机锡试剂使用量降为2倍物质的量时,收率仍维持在80%左右,与使用6倍物质的量时的收率相当;但当锡试剂量进一步降低为1.5倍物质的量时,化合物9的收率即会大大下降。我们最终选择使用2倍物质的量的三丁基乙烯基锡作为乙烯基试剂,该比例在保证转化率的同时降低了有机锡试剂的用量,降低了实验成本,减少了环境污染。

表2 有机锡试剂用量对化合物9收率的影响
2.3 脱氧核苷羟基保护基团的选择与脱除
在目标分子的合成过程中需要对脱氧核糖中3′-和5′-端的两个羟基进行保护来避免副反应的发生,常见的保护方法有酯类保护(乙酸酯保护,碳酸环酯保护和对甲基苯甲酸甲酯保护等)和醚类保护(硅醚保护,苄醚保护和烷氧基甲基醚保护等)[16-18]。酯类保护基团的脱除多通过碱性水解来实现,而硅醚类保护基团多通过含氟的季铵盐,如四烷基氟化铵TBAF(四丁基氟化铵)来脱除。Shigeki Sasaki课题组选用了TBDMS(叔丁基二甲基硅醚)来作为脱氧核苷中羟基的保护基团,并使用TBAF来进行脱保护。我们在重复实验时发现, TBAF是一种强亲核氟化剂[19],处理不干净对后续的反应会产生极大的干扰,并且其脱保护时间很长(24 h以上)。因此本文选择了含有Tol保护基团(对甲基苯甲酸甲酯)来对脱氧戊糖环的羟基进行保护,使用氨气的饱和甲醇溶液来切除保护基团。论文对实验的反应时间以及温度进行了探究,来探索脱保护的最佳条件。表3中的实验结果证实切除保护基团的氨解反应在温度80 ℃,时间2 h可以得到较高的收率。脱除保护基团的化合物6在乙酸乙酯溶剂中进行结晶即得到高纯度的结晶。
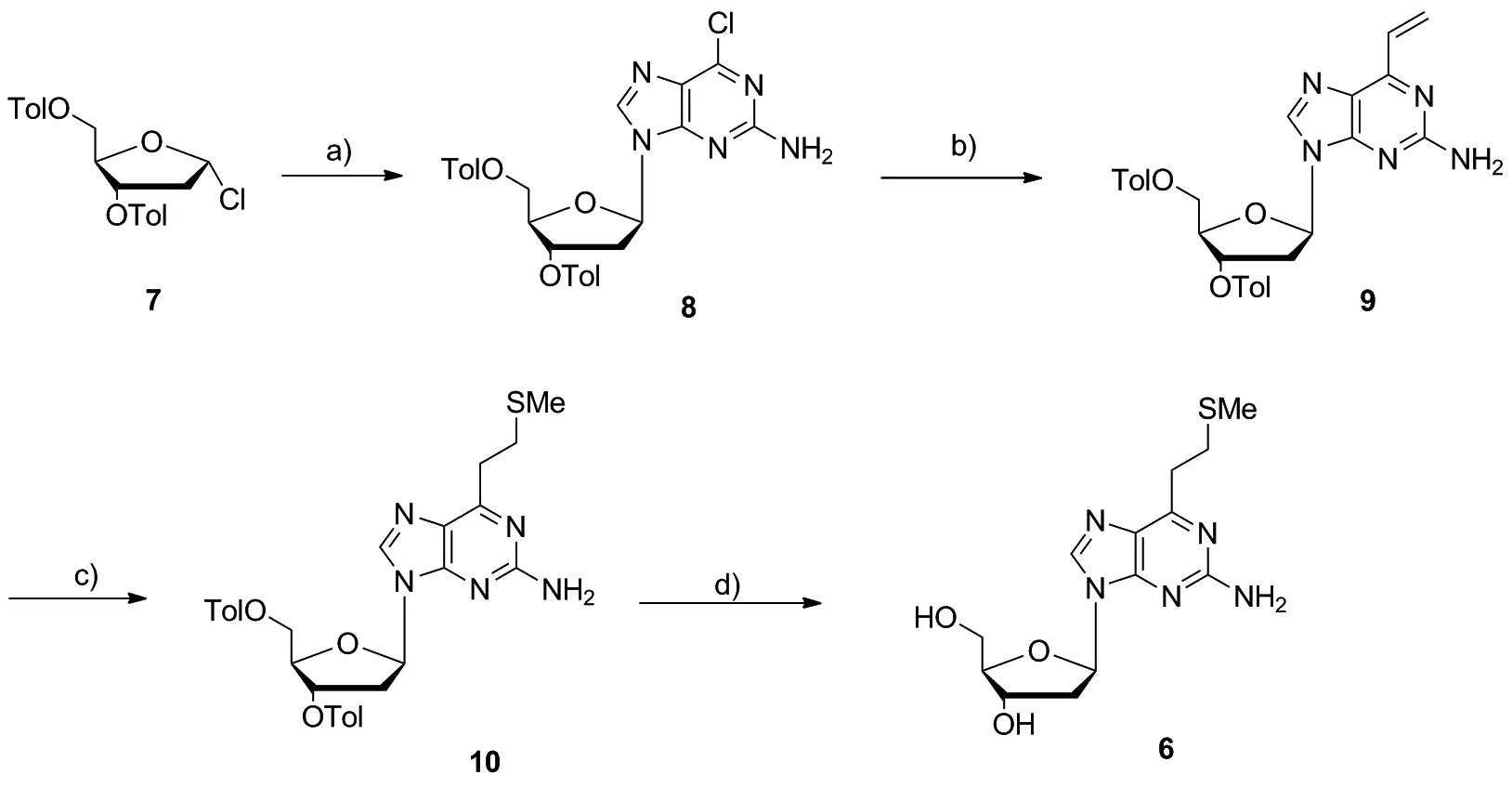
Scheme 2

表3 氨解反应时间及温度对化合物6收率的影响
报道了一种合成可交联嘌呤类脱氧核苷类似物的新方法。该方法以商业化的氯糖为起始原料,经过4步有机合成制备了可交联鸟嘌呤脱氧核苷。通过优化反应条件,在降低实验成本的同时减少了环境的污染,并改进了脱保护的条件和最终产品的纯化方法。
