HPLC 法测定药用辅料L(+)- 酒石酸中的D(-)-酒石酸含量
2022-04-26贾玉荣尤昆
贾玉荣, 尤昆
(江苏正大清江制药有限公司,江苏南京 210046)
1 引言
酒石酸有两个相同的手性碳原子,有三种立体异物体(左旋体(L-酒石酸)、右旋体(D-酒石酸)、 内消旋体)。 左旋酒石酸与右旋酒石酸为对映异构体。 天然存在的酒石酸都是右旋体。 右旋型酒石酸以游离的钾盐、钙盐、镁盐的形态广泛分布于高等植物中,特别是多存在于果实和叶中。 在制造葡萄酒时,会沉积大量酒石(氢钾盐)。 另外,在霉菌和地衣类中也常见到它的存在。 等量左旋体和右旋体的混合物,其旋光性相互抵消,组成外消旋体。 外消旋型酒石酸又称为葡萄酸。 葡萄汁或其他浆果汁在发酵制酒过程中,析出不溶于乙醇和水的右旋酒石酸的酸式钾盐沉积于酒桶壁上,称为酒石,酒石酸的名称就是由此而来。 左旋酒石酸可由外消旋体拆分获得,也存在于马里的羊蹄甲的果实和树叶中。
L(+)-酒石酸广泛用作饮料和其他食品的酸味剂,用于葡萄酒、软饮料、糖果、面包、某些胶状甜食。 利用其光学活性,作为化学拆分剂,用于制造抗结核病药物中间体DL-氨基丁醇的拆分;还可以作为手性原料用于酒石酸衍生物的合成;利用其酸性, 作为涤纶织物树脂整理的催化剂,谷维素生产的pH 调节剂。
L(+)-酒石酸同时为药用辅料,是DL-酒石酸拆分获得的。 其为注射剂中常用的pH 调节剂。中国药典2020 版[1]收录了L(+)-酒石酸的质量标准,目前文献报道的酒石酸含量测定方法有毛细管电泳法[2]、单柱阴离子色谱法[3]、电位滴定法[4]、高效液相色谱法[5,6]、原子吸收光谱法法[7]、分光光度法[8,9],其中高效液相色谱法具有简便快速、灵敏度高的优点,为目前测定有机酸类化合物最有效的手段[10,11]。但均未见文献对其异构体D(-)-酒石酸的含量测定进行报道。 本文采用HPLC 法测定L(+)-酒石酸中的D(-)-酒石酸,为提高该药用辅料的质量标准提供参考。
2 材料与方法
2.1 仪器、试剂与材料
高效液相色谱仪(美国Thermo Fisher 公司,UltiMate 3000),XS205DU 型电子分析天平(感量为0.01mg, 瑞 士METTER TOLEDO 公 司),KH7200B 型超声波清洗器(昆山禾创超声仪器有限公司);乙醇(色谱纯,默克公司);正己烷(色谱纯,默克公司);三氟乙酸(色谱纯,阿拉丁);纯化水(杭州娃哈哈有限公司);标准品D(-)-酒石酸(厂家:阿拉丁,含量99.5%);标准品L(+)-酒石酸(厂家:阿拉丁,含量99.1%);药用辅料L(+)-酒石酸(贵州欣紫鸿药用辅料有限公司)。
2.2 实验方法
2.2.1 标准品储备溶液的制备
分别精密称取D(-)-酒石酸对照品10.27mg、L(+)-酒石酸对照品10.33mg,置不同100mL 量瓶中,加乙醇溶解并定容至刻度,分别作为D(-)-酒石酸和L(+)-酒石酸标准品储备液。
2.2.2 标准曲线溶液的制备
精密吸取D(-)-酒石酸和L(+)-酒石酸标准品各0.4mL、0.8mL、1.0mL、1.2mL、1.6mL、2.0mL, 分别置10mL 量瓶中,加乙醇稀释至刻度,摇匀,即得。
2.2.3 样品前处理
精密称取本品25mg,置25mL 量瓶中,加适量乙醇,超声处理(功率300W,频率50kHz)5min,放冷,加乙醇至刻度,摇匀,用0.45μm 微孔滤膜滤过,取续滤液作为供试品溶液。
2.2.4 液相色谱条件
色 谱 柱:CHIRALPAK IC (250mm ×4.6mm,5μm); 柱温:25℃; 流速0.5 mL·min-1; 进样量:10μL;检测波长:210nm;以乙醇-正己烷-三氟乙酸(200∶800∶1)为流动相进行等度洗脱,运行时间为30min。 典型色谱图见图1。
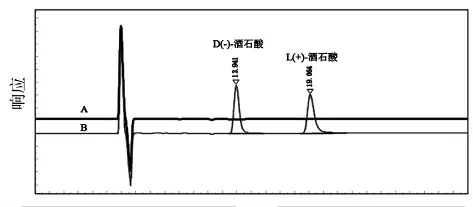
图1 A-空白溶剂;B-混合标准品溶液
3 结果与分析
3.1 线性关系
取配制好的标准曲线溶液, 分别进样10μL,按 “2.2.4” 色谱条件下测定峰面积。 以峰面积Y 为纵坐标,以标准曲线溶液浓度X 为横坐标进行线性回归,各组分的回归方程及相关系数见表1。 可见该方法再实验范围内线性关系良好。

表1 各组分线性关系测定结果
3.2 检测限、定量限
以信噪比为3 计算检出限,信噪比为10 计算定量限,测得D(-)-酒石酸、L(+)-酒石酸的检测限 分 别 为27.626ng、26.694ng, 定 量 限 分 别 为41.094ng、41.302ng。 该方法灵敏度高。
3.3 精密度
取标准曲线溶液(浓度水平3), 连续进样6次,结果测得D(-)-酒石酸、L(+)-酒石酸峰面积的RSD 分别为0.35%和0.28%,精密度结果见表2。
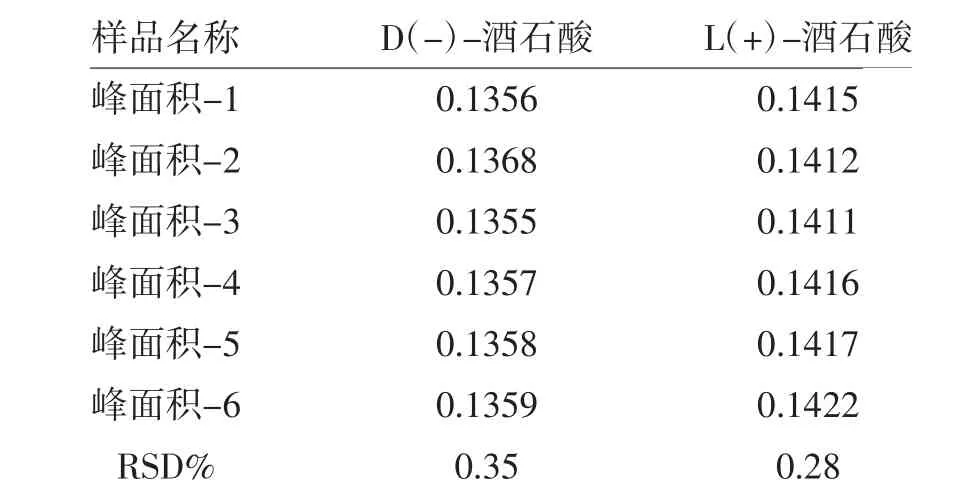
表2 精密度测定结果
3.4 重复性试验
取样品,平行试验6 份,结果D(-)-酒石酸的检出量为0.02%,RSD 为0.87%。 表明本方法重现性良好,结果见表3。

表3 重复性测定结果
3.5 中间精密度试验
取样品,由不同人员、在不同时间、采用不同仪器,平行试验6 份,结果D(-)-酒石酸的检出量为0.02%,与重复性6 份样品测定结果基本一致,12 份样品D(-)-酒石酸含量的RSD 为1.03%,方法的中间精密度良好,结果见表4。

表4 重复性测定结果
3.6 干扰试验
取空白溶剂-乙醇,按 “2.2.4” 色谱条件进样,空白溶剂在D(-)-酒石酸和L(+)-酒石酸相同保留时间附近无杂质峰干扰。
3.7 方法回收率
取9 份同批号样品约10mg(批号:Y20081001),精密称定,置10mL 量瓶中,每3 份分别精密加入L(+)- 酒石酸标准品储备溶液0.50mL、1.00mL、1.50mL,依法测定,结果D(-)-酒石酸回收率范围为99.6%~100.4%,RSD 为0.27%,回收率结果见表5。

表5 回收率测定结果
3.8 溶液稳定性
取供试品溶液, 分别于0h、2h、4h、8h、12h、24h 进样,依法测定,结果表明该供试品溶液室温放置24h 内,D(-)-酒石酸含量基本一致,峰面积的RSD 为1.47%,且无新杂质生成,说明供试品溶液在24h 内稳定。 结果见表6。

表6 溶液稳定性结果结果
3.9 耐用性
适当改变流速(0.5±0.1mL/min)、柱温(25±2℃)、流动相比例(205∶795,195∶805),考察系统适用性溶液中D(-)-酒石酸与L(+)-酒石酸之间的分离情况。L(+)-酒石酸与其异构体峰的分离度均大于1.5,符合要求,表明该分析方法的耐用性良好。 结果见表7。

表7 耐用性试验结果
3.10 样品测定
取3 批样品,每批样品平行测定2 份,结果D(-)-酒石酸含量分别为0.015%、0.021%、0.027%。
4 讨论
4.1 流动相的选择
D(-)-酒石酸和L(+)-酒石酸为构型异构体,需采用手性柱进行分离。 选用正相体系通用的流动相正己烷-乙醇进行流动相筛选, 通过添加不同种类的酸性调节剂改善峰型, 主要考察了甲酸、乙酸及三氟乙酸。 结果以正己烷-乙醇-三氟乙酸为流动相时,峰形好,分离度大于5.0,且空白溶剂及其他杂质无干扰。
4.2 波长的选择
对D(-)-酒石酸进行了紫外全波长扫描,结果显示该成分无最大吸收波长, 仅有末端吸收,因此选择210nm 作为检测波长。 在该检测波长下,D(-)-酒石酸的检测限较低,灵敏度好。
4.3 流速的选择
采用手性柱进行分离, 对流速进行了筛选。结果表明当流速为0.5mL·min-1时,D(-)-酒石酸和L(+)-酒石酸的分离度合适,柱压较低。流速越低,消耗的流动相越少,柱压也越低,可以节约试验成本,同时减少对仪器的损耗。
4.4 柱温的选择
柱温过低会导致出峰时间延长,柱压力增大;柱温过高会导致柱流失。 常规采用25℃或者30℃进行液相色谱分析。 考虑到实验室的环境控制一般都在25℃左右,因此选择25℃作为柱温。 结果表明该柱温条件下,出峰时间及分离度均较适宜。
