电感耦合等离子体原子发射光谱法和电感耦合等离子体质谱法检测药品中元素杂质的研究进展
2022-04-24尹利辉许明哲
朱 俐,赵 瑜,尹利辉,许明哲
(化学药品质量研究与评价重点实验室,中国食品药品检定研究院,北京 102629)
保证药品安全、有效、质量可控是药品研发和评价应遵循的基本原则,其中对药品进行质量控制是保证药品安全有效的基础和前提,也是确保药物质量最重要的挑战之一[1-2]。药品中杂质分析是药品质量控制的重要组成部分,近年来,由于对药物中杂质限度要求的不断提高,许多药典杂质分析方法已经修订或更新。药品中的元素杂质不仅影响药物的稳定性,而且可能存在潜在毒性,进而可能引起健康风险,因此必须严格控制药品中元素杂质的含量。
早期各国药典对元素杂质的控制主要是重金属检查法,其原理是基于有毒元素(大约为10种)与硫代乙酰胺生成有色硫化物沉淀,视觉上与标准溶液的颜色进行比较,以确定是否超出重金属限度。该方法基于总量控制模式(除砷盐检查外),未对单个元素杂质定量分析,方法的专属性和灵敏度均较低,无法满足各国对药品安全性控制的发展要求。2008年,欧洲药品管理局(EMEA,现简写为EMA)正式颁布了《金属催化剂或金属试剂残留限度指南》,首次制定了药物中14种金属元素杂质的限度要求。2014 年,国际人用药品注册技术协调会(ICH)Q3D 元素杂质指南提出了药品中元素杂质的分类和控制要求。各国药典对元素杂质的控制也越来越严格,欧洲药典(EP)从9.0 版起,删除了原通则2.4.8-重金属检查法,美国药典(USP)从第35版第二增补本起,增订通则〈232〉元素杂质-限度和〈233〉元素杂质-方法,2018年后停用了〈231〉-重金属检查法,目前已修订为与ICH Q3D 一致。日本药典(JP)作为ICH 的发起成员之一,日本对药物中元素杂质控制执行ICH Q3D 的相关要求。而在《中华人民共和国药典》(2020 年版)(简称ChP 2020)中对元素杂质的检测仍采用重金属限度检查法,但收载了0411电感耦合等离子体原子发射光谱法(ICP-AES)和0412 电感耦合等离子体质谱法(ICP-MS),未新增与元素杂质相关的技术要求,修订〈9102〉药品杂质分析指导原则:明确无机杂质参照ICH Q3D 进行研究并确定检查项目。原子吸收光谱法(AAS)虽然可以定量检测药品中元素杂质,但由于该方法耗时、不能高通量检测等,仍需要开发新的元素杂质的分析方法克服其缺点,比如更高通量、更低试剂消耗和更高的灵敏度的方法[3-4]。ICPAES和ICP-MS由于具有快速和多元素分析等优点[5-8],被USP〈233〉推荐为元素杂质的检测方法。
1 药品中的元素杂质
文献中广泛使用的“重金属”指的是与污染和潜在毒性相关的金属、类金属和一些非金属[9]。但实际上“重金属”具有不同的含义,并且与化学或毒理学没有基本的相关性。这种用法意味着某种元素(有机和/或无机)的所有化合物都具有相同的物理、化学、生物学和毒理学性质,这对于大多数元素而言并非如此。ChP 2020中的重金属限度检查法中的重金属是指在试验条件下能与硫代乙酰胺或硫化钠试液作用而显色的金属杂质,如银、铅、汞、铜、镉、铋、锑、锡、镍、锌等,但某些具有毒理学意义的元素(如通常用作催化剂的铂族元素)未涵盖在内。
药物制剂组成复杂,包含的无机元素种类较多,金属杂质仅能代表金属类的杂质,不能涵盖非金属(例如砷)的杂质,ICH 专家工作组在2013 年将Q3D 的金属杂质(metal impurities)修订为元素杂质(elemental impurities),并基于风险评估的原则对元素杂质进行控制。根据元素的毒性及其在药品中出现的概率(以硅为参照,若该元素的自然丰度小于硅的百万分之一,则归为低天然丰度元素),将元素杂质分为以下几类。
1类元素:是对人体有致癌性、遗传毒性、神经毒性或肝肾毒性的元素,在生产中禁用或限制使用,通常来源于常见物料的引入(如矿物质辅料等)。所有给药途径中,此类别元素均需进行评估,包括砷、镉、汞、铅等4种元素。
2类元素:是给药途径依赖型的有害元素,根据元素在药品中出现的可能性,分为2A 和2B类。
2A 类元素:较1类元素的毒性低,但出现在药品中的概率较高,所有给药途径中,此类别的元素均需进行评估,包括钴、镍、钒等3种元素。
2B类元素:此类元素的自然丰度较低,与其他物料潜在共生的可能性也较低,在药品中出现的概率也较低。除非在原料药、辅料或其他药品组分的生产中有意添加,此类别的元素一般不需考察和控制,包括银、金、铱、锇、铅、铂、铑、钌、硒和铊等10种元素。
3类元素:此类元素口服给药时的每日允许暴露量(PDE)500μg·d-1,毒性较低。除非有意添加,口服给药时一般无需考虑此类元素杂质的残留风险,但在吸入和注射给药时,应对此类元素的风险进行评估。如注射给药时,需同时考察锂、锑、铜的残留;吸入给药时,需同时考察上述全部3类元素的残留,包括钡、铬、铜、锂、钼、锑和锡等7种元素。
其他类元素:该类元素PDE 值尚未建立,其固有毒性低或区域监管法规存在差异。此类元素建议按各地区域监管法规要求控制。对特意加入到制剂中的有治疗价值的元素,ICH Q3D 未将其列为元素杂质。
ICH Q3D 为口服、注射和吸入制剂中的24种元素杂质建立PDE值,重点强调药物生产过程中有意添加使用的所有元素,应独立于给药途径和相应类别进行评估[10]。目前USP、EP 都已经明确对元素杂质的控制要与Q3D 保持一致。特别需要指出ICH Q3D 提及的元素杂质不包括铝,但肠外营养注射液需要测定铝,因为铝污染可能导致接受肠胃外营养治疗的患者组织中铝积累,特别是新生儿和肾功能受损的患者。因此,对于这些药品,还应对铝进行风险评估[11]。
2 污染源
药物中的杂质取决于合成工艺,杂质可能来源于合成路线、盐析方法和萃取过程中使用的溶剂[12]。在原料药生产过程中添加的元素(例如催化剂)可能无法完全除去,应考虑这些杂质的最大容许量,有文献报道采用AAS或ICP-MS测定药物中的铱、锇、钯、铂、铑、钌和钨等催化剂残留量[6,13-15]。元素杂质还可能来源于使用的设备以及与工艺中所有可接触的金属表面,如果表面材料不耐腐蚀,也会引入元素杂质镍等,比如玻璃[16]中的铝、橡胶[17-18]和活性炭[19]材料中的锌,以及玻璃容器封闭系统中[20]铝、砷和钨的污染。通常,原料中最易引入元素杂质,终产品中可能会存在来自原料、试剂或辅料的一些元素杂质。元素杂质的另一个来源与使用的水净化系统有关,从原料合成到成品的许多过程中都使用水,因此在所有药典中都强烈建议测定水中的元素杂质。
3 等离子体光谱技术
等离子体技术的特征是高温(6 000~10 000 K)可以使大多数元素雾化和离子化。电感耦合等离子体(ICP)的仪器中用于产生等离子体的气体是氩气,也可用氦气和氮气,但非常少,因为大多数元素在氩气等离子体环境中,只能电离成单电荷离子,进而可以很容易地分离并加以检测。目前检测常用元素的ICP-AES和ICP-MS是USP〈233〉推荐的用于药物中元素杂质检测的方法。
3.1 ICP-AES和ICP-MS的总体比较
相比较于AAS,ICP-AES和ICP-MS的主要优势就是可以多元素同时分析。虽然X 荧光光谱法也可多元素同时分析,但其精度远远低于ICP 技术。最初对药物中痕量元素杂质检测常采用AAS,但随着人们对药品中元素杂质的重视,使用基于ICP-AES和ICP-MS的高通量方法测定药物中的元素杂质逐渐增加,预计ICP-AES和ICP-MS将成为该领域的首选方法。2008年,EMA[21]和USP[22]发布两份重要文件均涉及元素杂质的评估,这些文件表明了根据给药方式和PDE 值为每个元素设立含量限值的重要性。考虑到要测定元素种类众多,分析工作者认为ICP-AES和ICP-MS是最适用的元素杂质检测的方法[22]。ICP-AES 和ICP-MS 还有一个较大的优点就是具有相对较低的检出限,ICP-AES通常可达到μg·L-1,而ICP-MS可达到ng·L-1。ICP-AES和ICP-MS还具有线性范围宽的优点,通常线性范围可扩展到106~109[23-24]。由于低于190 nm 的真空紫外光容易被氧气和水汽吸收,因此以前开发的大多数ICP-AES都不能应用于125~190 nm,但许多元素(比如砷、硒、磷、硫和卤素等元素)的灵敏线均在125~190 nm 内,随着仪器的发展,吹扫和抽真空系统的应用使得某些电感耦合等离子体原子发射光谱仪可以在190 nm 以下(真空紫外线区域)检测砷、硒、磷、硫和卤素等元素。ICP-MS同样也可以实现强大的检测功能,ICP-MS多采用四极杆质量分析器,以扇形磁铁代替四极杆形成高分辨ICP-MS技术已经非常成熟[25],以多接收检测器代替单接收检测器对同位素比值精密分析非常重要,也是ICP-MS的未来发展趋势[26-27]。
3.2 ICP-AES和ICP-MS技术的干扰
尽管ICP-AES和ICP-MS有很多优点,但ICPAES和ICP-MS对元素杂质检测时会有光谱和非光谱干扰,通常ICP-MS比ICP-AES更容易受到干扰。由于ICP的激发能力很强,几乎每一种存在于ICP中或引入ICP 中的物质都会发射出丰富的谱线,从而产生大量的光谱干扰。非光谱干扰会导致信号抑制或增强,通常会在样品溶液与标准溶液的成分非常不同的情况下发生,从而导致结果有偏差。因此,在元素分析之前应充分了解药品的成分,以识别潜在的干扰,分析时应着重考虑溶剂类型、溶解盐、残留碳和消化液的酸度。大多数药物活性成分(API)是有机物,其中一些API除了碳和氢以外还包括其他元素,如氯、氮、氧和硫。此外,酸性和碱性原料药通常以盐的形式存在,几乎有20%的API是盐酸盐,在ICP-MS测定某些元素时会有氯的光谱干扰。BARIN 等[5]发现,含氯100 mg·L-1的水溶液对51V+、52Cr+、53Cr+和75As+的检测存在干扰。肠外制剂和滴眼液通常用氯化钠溶液调节渗透压,因此对其中元素杂质的检测可能会受到氯的干扰。ICP-MS测定中,硫会干扰65Cu+的测定,而磷会影响63Cu+和65Cu+的测定[5],化学结构中包含硫或磷的药物(如磺酰胺和噻嗪类)会干扰检测,还需要注意药物中的赋形剂(如磷酸钙)也会干扰检测。还有很多API中含有碱金属和碱土金属元素(如钠、钾、钙和镁)的盐,这类API因为含有大量容易离子化的元素(EIE),采用ICP-AES和ICP-MS检测时会产生极大的干扰。对于ICP-MS,低电离势元素会产生较大的信号抑制,并且抑制信号的强度随元素的质量增加而增加,这与空间电荷效应有关[28];对于ICP-AES,EIE和非EIE会影响离子化、雾化、检测等整个分析过程[29]。
溶液中的碳含量也是一个重要的参数,也就是总固体溶解量(TDS)。碳源可以是API、赋形剂,甚至是样品制备过程中未消解的残留碳。高含量溶解碳会因为在溶液中存在的形式不同而产生不同的干扰[30-31]。ICP-MS 分析碳含量较高的溶液时,可以观察到非光谱和光谱干扰。在等离子体中形成多原子离子以及在采样锥、截取锥和离子透镜系统沉积,会导致等离子体特性和离子分布的改变,这些变化主要是由分析物和含碳带电物质之间的电荷转移引起的[32]。AGATEMOR 等[32]讨论了ICP-MS的基质效应,发现当碳质量浓度高于750 mg·L-1时,信号强度增加,并对52Cr+(由40Ar12C+产生)和53Cr+(由40Ar13C+产生)产生光谱干扰;碳质量浓度降低至250 mg·L-1时,对52Cr+和53Cr+也会产生干扰;碳质量浓度增加至2 000 mg·L-1时,对60Ni+检测会产生干扰,如果用碰撞反应池可减少干扰程度[6]。ICP-MS 还存在多原子干扰,比如,40Ar12C+对52Cr+,40Ar13C+对53Cr+以 及12C16O16O16O+对60Ni+的干扰。根据经验,消化方法的效率必须足够高,消化液中的碳质量浓度低于250 mg·L-1时可把干扰降到最低。对于药物中某些含量非常低的元素的检测,则需要考虑增加TDS,使检测溶液的含量大于检出限,但同时也要考虑如何消除多原子干扰,以保证检测的准确度。
采用ICP-AES和ICP-MS检测,还需要控制溶液的酸度,避免干扰样品引入(比如产生气溶胶)和等离子体特性的改变。对于ICP-MS,要注意酸种类的使用,如盐酸对检测砷、硫酸对检测铜均会出现光谱干扰。样品用无机酸直接溶解或消解后,溶解的酸度可能会影响进一步分析,此时需进行赶酸操作,既可以降低干扰也可以保护仪器。样品制备过程中使用有机溶剂也容易产生光谱干扰[30,33-34],减少或消除ICP-MS中的光谱干扰(主要是多原子干扰)的最佳选择是使用碰撞反应池[35]。在优化的流量下,单一使用氦碰撞气时,分析物信号强度有可能会更低而影响检测的准确度[36],可添加氢气消除Ar O+的干扰[37];用氦-氨混合气可以检测左旋多巴、二磷酸伯氨喹、盐酸普萘洛尔和磺胺甲口恶唑[6]、降压片剂[38]和肠外营养液[39]中的元素杂质,避免对铬、铜、锰和钒的多原子干扰。
3.3 ICP-AES和ICP-MS的非常规使用
依据样品制备的方法,进样系统可以使用常规的气动雾化、超声雾化、化学气相生成(CVG)和其他方式。流动注射(FI)系统与ICP-MS结合使用可实现高通量和低消耗,从而可测定API中的元素杂质[26,40]。CVG 是在电感耦合等离子体原子发射光谱仪或电感耦合等离子体质谱仪中引入分析物的一种方式,可以从基质中高效分离出分析物,减少干扰并降低检出限[41]。CVG-ICP-MS 可以检测汞元素[42],降低汞的记忆效应,与常规雾化系统相比更加灵敏。还有一种膜去溶剂化装置,该装置可以连接到雾化系统,将有机溶剂引入ICP 仪中,使得ICP-AES和ICP-MS技术可以直接分析有机溶剂的样品。TU 等[43]用N,N-二甲基甲酰胺作为通用有机稀释剂,将样品制备简化为稀释-加样程序,用膜去溶剂化装置检测药品中15种元素,灵敏度提高了2~10倍。
激光烧蚀(LA)[44]和电热蒸发(ETV)[45]系统可以将固体和半固体样品引入等离子体,LA-ICPMS不需要制备样品就可快速获得分析信号[46],是药物中元素杂质筛查的有力工具,但该方法需要与待测样品基体匹配的标准物质以及纳克级的样品量,因此可能会存在定量准确度和样品代表性问题。ETV-ICP-AES[44]和ETV-ICP-MS[38]也可以直接检测固体药物中的元素杂质,与LA 系统相比,ETV 系统可以使用相对多的样品量(最多可以是2.5 mg),该方法的另一个优点是分析物汽化前可通过热解步骤去除基质,从而可以使用参比水溶液进行校准,过程中可能需要使用其他气体(例如氟利昂)[44]或化学改性剂溶液[38]以改善分析物的蒸发行为,但是由于热解步骤中的损失[44],有些元素无法进行准确定量。
4 样品制备
文献报道常用的ICP-AES 和ICP-MS 检测药物中元素杂质的样品制备方法有直接溶解和消解等,上述方法都是USP〈233〉推荐的样品制备方法,具体见表1。直接溶解是首选的样品制备方法,简单、容易操作,要求样品溶于水或者稀酸溶液,但有些API难溶于水或者酸性水溶液(如头孢曲松),并且药物制剂中还包含很多难溶的辅料(如黏合剂、着色剂、颜料等),这时需要采用高压微波消解法制备样品。无论选择什么样的方法,样品制备最重要的是防止待测物的损失。由于药物样品的多样性,样品制备具体方法的选择取决于样品的性质[47]。需要注意的是,选择溶剂溶解或消解样品时,需考虑样品的化学稳定性和挥发性,还应考虑待测元素的稳定性,避免目标元素的损失,比如对汞和锇等元素进行分析时,需在消解时加入稳定剂,以保证其稳定。

表1 文献中ICP-AES和ICP-MS检测药物中元素杂质的样品制备方法Tab.1 Sample preparation methods for ICP-AES and ICP-MS determination of elemental impurities in drugs in the literature
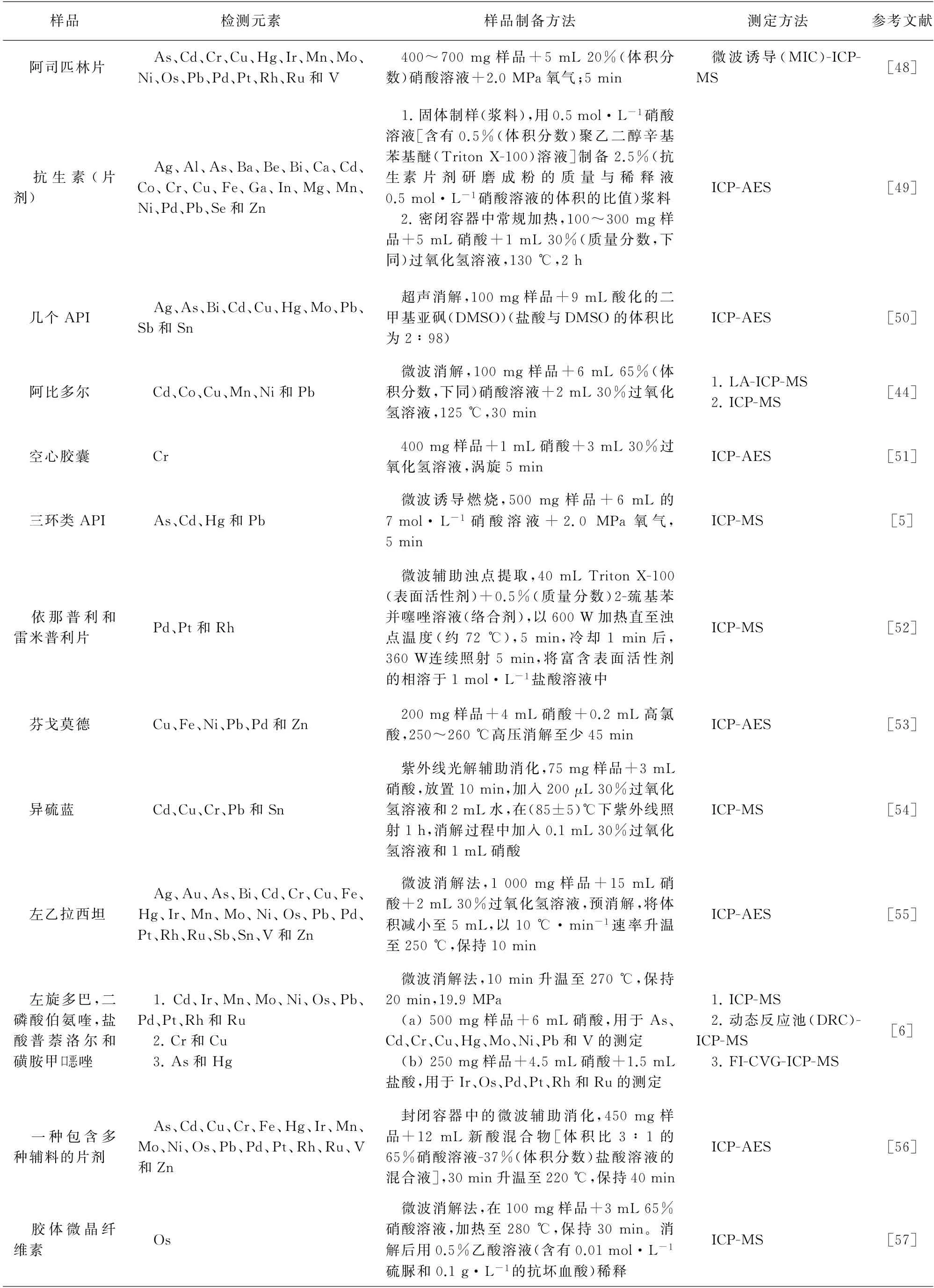
表1 (续)
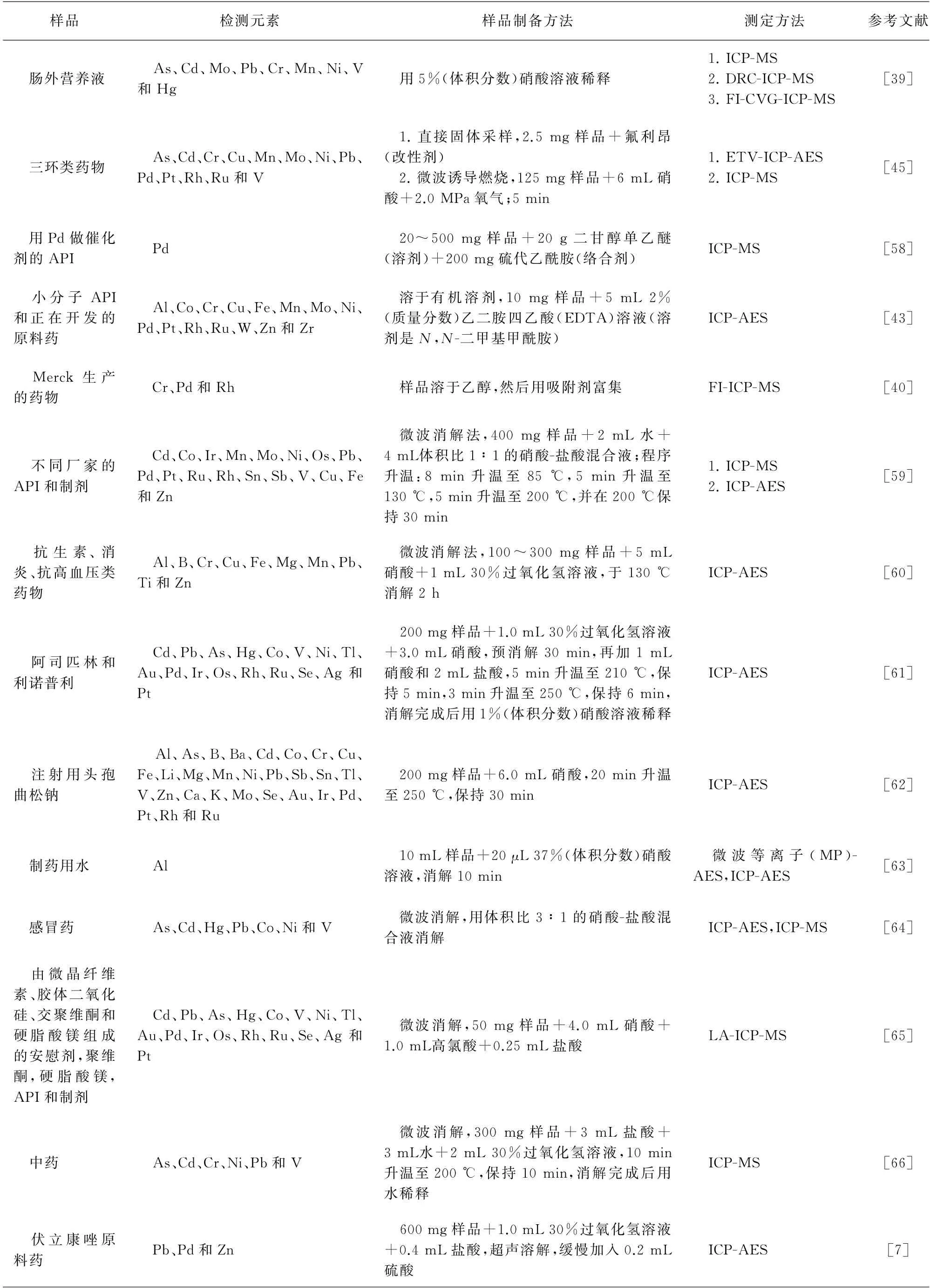
表1 (续)

表1 (续)
4.1 直接溶解
如果样品溶解于水,直接溶解方法通常用稀酸的水溶液作为溶剂,样品量在10~100 mg内可以最大程度地减少碳含量引起的干扰。需要注意的是,由于样品量的限制可能会造成含量很低的元素低于检出限,采用FI系统获取瞬时信号[25,40]可以降低直接溶解法的碳含量。不溶于水的样品可以采用有机溶剂溶解,其检测仪器需要采用膜去溶剂化装置或者雾化器通入氧气。N,N-二甲基甲酰胺[42]、二乙二醇单乙醚(DGME)[58]、乙醇[40]和2-丁氧基乙醇[70]都可以作为API的有机溶剂。
药品引入的元素杂质大部分是API合成过程中加入的重金属催化剂,这些元素杂质与API通常不发生化学键合,因此可以采用浊点提取(CPE)[52]和超声辅助提取[50]元素杂质。CPE 通常用于从液体样品中提取和预浓缩待测元素,一般用含表面活性剂和配体的混合物提取元素杂质,元素杂质与配体结合后转移至表面活性剂相。对于固体基质,超声辅助提取可提高提取效率。与传统的固液萃取相比,超声辅助提取与CPE 的优点是操作简单,但必须使用合适的配体以确保回收率[52]。
与微波消解方法相比,直接溶解样品的方法非常简单,操作步骤少,减少了待测元素的损失,这对于快速筛查和常规监测元素杂质非常重要。
4.2 消解
湿法消解是用氧化性的强酸通过加热或微波对样品进行消解,该方法是最常见的基质消解方法。硝酸是消解常用的酸,因为硝酸的氧化能力适当,并且可通过蒸馏纯化获得高纯度试剂,还有硝酸盐一般都溶于水。消解液的选择是湿法消解的重要条件,消解液必须确保分析物完全消解并以稳定的形式存在于溶液中。
大多数药品只用硝酸就可以消解[71],或者加入过氧化氢,过氧化氢用量需要在方法中优化,如体积比7.5∶1[55],5∶1[49]的硝酸-过氧化氢混合液都可以用来消解药品。根据样品基质,还可以用硝酸与盐酸、氢氟酸、高氯酸、磷酸、硫酸的混合酸消解[72-73],当样品中含有二氧化硅(SiO2)、二氧化钛(TiO2)或者滑石粉[Mg3Si4O10(OH)2]时,需要用氢氟酸消解样品[60,74]。
还有一些元素杂质(主要是汞、铱、锇、钯、铂、铑和钌)的消解液选择至关重要。用体积比为3∶1的盐酸-硝酸混合酸消解样品,在消解后的溶液中,锇在6 h之后不稳定[57],可用含1%(体积分数)盐酸溶液[75]或0.01 mol·L-1硫脲(硫脲溶于0.1 g·L-1抗坏血酸)的0.009 mmol·L-1溴酸钾溶液作为络合剂来稳定溶液中的锇[25,57]。对于汞,以5%(体积分数)盐酸溶液作为络合剂可稳定汞并且能够降低雾化系统的记忆效应,进而获得良好的回收率[25],Au3+也可以起到与盐酸类似的作用,可以稳定Hg2+和降低记忆效应[56]。根据经验,要想准确测定汞、锇、钯和铂族元素不太容易,即便采用文献报道的方法,也不一定获得良好的结果,需要仔细地优化条件并评估方法的准确性。表1列出了文献使用湿法消解的简要条件(样品量、消解液组成及加热程序),可作为优化制备方法的参考。
有些API直接湿法消解比较困难,则可以采用高温高压微波消解样品,在密闭容器中微波消解不仅可以提高消解效率而且可以最大程度减少损失和污染,检测挥发性元素最好采用密闭容器微波消解,并且要加入稳定剂。密闭的消解罐材料一般是石英、聚四氟乙烯或其他惰性材料,消解温度可在200~280 ℃内[68],压力可达8.0 MPa[72]。
湿法消解的关键是要确保样品完全溶解并且有较低的碳残留(RCC)[6],样品量通常最大为500 mg,但也有用高于1 g的样品量[72-73]。最终样品溶液的酸度应该与标准溶液保持一致,标准溶液的酸度较高时,会抑制分析信号。
5 结论
Ch P中重金属限度检查法的局限性显而易见,目前国家药品监督管理局已经要求对还未上市或在申报的药品全面执行ICH Q3D。药物中元素杂质种类较多,元素杂质检测工作艰巨而复杂,ICP-AES和ICP-MS因其独特优势而成为USP〈233〉推荐的元素杂质检测方法,并在药品行业得到广泛的使用。ICP-AES和ICP-MS检测药物中元素杂质时,样品制备可以直接溶解在水、稀酸溶液或者有机溶剂中,还可以用敞开式湿法消解和密闭容器中微波消解,优化的样品制备方法为药品中元素杂质检测提供了良好的检出限。需要注意的是,对于不稳定或挥发性元素需要加入稳定剂才能获得可靠的分析结果。ICP-AES和ICP-MS均可使用ETV 或LA 来检测固体样品,几乎所有元素杂质均能获得良好的结果。但是由于API多种多样,药物有不同的剂型并且包含各种辅料和赋形剂,每种情况都会有不同的解决方案,要想开发一种通用的可以准确测定多种元素杂质的ICP-AES和ICP-MS方法或法定方法将会是药物分析方面的一项挑战。
