化学交联质谱技术的研究进展*
2022-04-24陈镇霖贺思敏
陈镇霖 曹 勇 贺思敏**
(1)中国科学院计算技术研究所,中国科学院智能信息处理重点实验室,北京100190;2)中国科学院大学,北京100049;3)北京生命科学研究所,北京102206)
化学交联质谱技术(chemical cross-linking coupled with mass spectrometry,CXMS)是解析蛋白质结构和研究蛋白质相互作用的重要工具。该技术利用化学交联剂将空间距离足够接近的两个氨基酸以共价键形式连接起来,然后通过质谱技术鉴定发生交联的两个氨基酸位点。由于交联剂臂长的限制,两个交联位点之间的距离不应超过交联剂的臂长,该距离信息将为蛋白质结构解析和相互作用研究提供重要的约束条件。相比于传统的蛋白质结构解析和相互作用研究技术,化学交联质谱技术具有分析速度快、通量高、对蛋白质样品的量和纯度要求低以及可捕获弱相互作用等优势[1]。
化学交联质谱技术自本世纪初提出以来[2],获得了学术界的广泛关注。截至2020 年底,有关该技术的研究论文已累计发表超过5 000 篇,累计被引超过15万次;特别是最近5年,每年发表的文章数量均超过了300 篇,被引次数均超过了1 万次(数据来源于Web of Knowledge, http://apps.webofknowledge.com/,2020 年12 月24 日),说明该技术持续成为领域的研究热点,具有重要的研究价值。一些综述对近几年的化学交联质谱技术进行了总结[3-6]。
经过数十年的发展,化学交联质谱技术在各个环节都取得了长足的进步。目前,一个典型的化学交联质谱技术的工作流程主要包含8 个步骤(图1),依次是:交联剂选择、交联反应、酶切、交联肽段富集、液质联用、交联肽段鉴定、质量控制和生物学应用。下面将按照化学交联质谱技术工作流程的先后顺序进行综述,重点介绍最近5年的研究进展,最后对该技术进行总结与展望。
1 交联剂
交联剂的结构通式通常由两个反应基团(reactive group) 和连接它们的交联臂(spacer arm)组成(图1a)。如果交联剂的两个反应基团相同,则称为同型的(homodimeric),否则称为异型的(heterodimeric)[7]。目前的交联剂设计已经模块化,通过组合不同的反应基团和交联臂,可以得到各具特色的新型交联剂。下面分别介绍具有不同反应基团和不同交联臂的交联剂。

Fig.1 The workflow of CXMS图1 化学交联质谱技术的工作流程
1.1 交联剂的反应基团
目前最常使用的反应基团是与氨基反应的琥珀酰 亚 胺 酯 基 团 (N-hydroxy succinimidyl 或sulfosuccinimidyl esters,NHS ester),它能特异地与赖氨酸K侧链的氨基或者蛋白质N端的氨基发生交联反应。有研究表明,在酸性pH 或交联剂浓度较高时,琥珀酰亚胺酯基团还可能和丝氨酸S、苏氨酸T或酪氨酸Y反应[8]。常见的使用琥珀酰亚胺酯 基 团 的 交 联 剂 有DSS[9]、BS3[10]、PIR[11-12]、DSBU[13]和DSSO[14]。
虽然琥珀酰亚胺酯基团的反应效率较高、反应副产物较少,但如果目标蛋白质所含赖氨酸数量较少时,使用这类交联剂无法得到足够多的交联位点信息。为了获得更多的交联位点信息,研究人员设计出了许多包含不同反应基团的交联剂,例如EDC[15]、DMTMM[16]和Diazoker[17]分别包含与羧基反应的二酰肼和重氮,可以与天冬氨酸D和谷氨酸E 反应;BMSO[18]包含与巯基反应的马来酰亚胺,可以与半胱氨酸C 反应;ArGO 和KArGO[19]包含与胍基反应的苯乙二醛,可以与精氨酸R反应。
更进一步,一些交联剂会使用非特异的反应基团,可以和数个甚至所有氨基酸发生交联反应,此类交联剂被称为非特异交联剂。例如使用磺酰氟的NHSF[20],可以与数个亲核氨基酸(组氨酸H、丝氨酸S、苏氨酸T、酪氨酸Y、赖氨酸K)反应;分别使用双吖丙啶和苯甲酮的sulfo-SDA[21]和sulfo-SBP[22],在紫外光照射下可以与任意氨基酸反应。虽然使用非特异的反应基团理论上可以获得更多的交联位点信息[23],但由于交联反应的产物更加复杂、交联肽段鉴定时的搜索空间更大,对交联肽段鉴定和质量控制都带来很大的挑战,因此现阶段此类交联剂使用并不多。
1.2 交联剂的交联臂
简单的交联臂通常是一条碳链(如DSS和BS3的交联臂)或聚乙二醇链(如Diazoker 和KArGO的交联臂)。改造交联臂,可以实现不同的功能,如添加可富集基团,可以富集交联肽段[11,24-26](见4.1);带上同位素标记,可以实现交联肽段的定量等功能[11,24,27-30]。
交联臂在质谱中是否可断裂对后续的质谱数据采集和交联肽段鉴定有很大影响。如果交联臂中包含比肽键键能更低的可断裂键,则质谱在断裂肽段主干的同时,也会断裂交联臂。当交联臂断裂之后,交联的两条肽段彼此分离,通过特征峰的分析,有可能得到两条肽段的质量,由此可将交联双肽鉴定问题转化为常规单肽鉴定,大大降低了交联肽段鉴定的复杂度(见6.2)。而为了既断裂交联臂,又断裂肽段主干,质谱数据采集方案也需要做相应调整(见5.3)。包含质谱可断裂交联臂的交联剂简称为质谱可断裂交联剂,由于其能降低交联肽段鉴定的复杂度,逐渐成为领域的研究热点。
目前常见的质谱可断裂交联剂的交联臂所含的可断裂键大致可分为两类,一类是C—S 键,例如类 似 于DSSO 的 一 系 列 交 联 剂[14,18,28,31]和CBDPS[29-30];另一类是C—N键,例如PIR[11-12]和DSBU[13]交联剂。除了质谱可断裂交联剂之外,还有一些交联剂的交联臂能通过紫外光照射[32]、化学反应[33]等方法进行断裂,此类交联剂往往在进质谱之前就已断裂,因此难以通过质谱鉴定发生交联的两条肽段。相比于质谱可断裂交联剂,紫外光、化学可断裂交联剂使用较少。
交联臂除了具有不同的功能基团之外,其自身的长度也是很重要的属性,称之为臂长。大多数交联剂的臂长在10~15 Å 之间,如DSS[9]、BS3[10]、DSBU[13]和DSSO[14];有少量交联剂的臂长接近于0,如EDC[15]、DMTMM[16]和CDI[34];还有少量交联剂的臂长很长,如PIR[11-12],臂长长达43 Å。一般来说,臂长越短,能形成的交联位点对越少,但由此获得的距离约束更紧,更适合用于蛋白质结构建模;在一定范围内,臂长越长,能形成的交联位点对越多,但由此获得的距离约束更松,更适合用于蛋白质相互作用研究[35-36]。
在模块化设计的思想指导下,产生了多功能交联 臂 的 交 联 剂, 例 如 交 联 剂PIR[11-12]和CBDPS[29-30]的交联臂同时具有可断裂、可富集和可同位素标记的功能。进一步,通过组合多功能交联臂和非特异的反应基团,甚至能够得到全能型的交联剂。2019年发表的pBVS交联剂就是一种全能型交联剂,它同时实现了可断裂、可富集和可与多种氨基酸反应的功能[37]。
甲醛是一种非常活泼的小分子,它既可以与多种氨基酸反应, 又可以与脱氧核糖核酸(deoxyribonucleic acid,DNA)反应,因此也可以作为一种交联剂。甲醛具有容易穿过细胞膜和核膜、交联反应可逆等优点,故常用于染色质免疫共沉淀(chromatin immunoprecipitation,ChIP)中固定DNA与蛋白质的相互作用[38]。此外,甲醛也被用于研究细胞内蛋白质与蛋白质的相互作用[39]。虽然甲醛具有很多的优点,但甲醛交联的作用机制尚不明确,很少有工作能够直接从甲醛交联样品中鉴定到交联肽段。2020 年,以色列希伯来大学的Kalisman 团队[40]推测,甲醛交联通过两步反应完成,且形成的交联剂质量为24 u、在质谱中容易断裂为两个12 u的修饰。因此,甲醛相当于一种非特异、可断裂的交联剂。
截至目前,虽然各种新型交联剂层出不穷,但最常使用的交联剂依然是DSS 和BS3[41],这一方面是因为这两种交联剂的反应特异性好、反应副产物少、数据分析方法成熟,另一方面是因为很多新型交联剂的设计理念虽好,但相应的质谱数据采集方案和交联肽段鉴定方法并不成熟,实际应用时效果并不理想。有研究人员呼吁,交联剂的设计不应该只是概念驱动(concept-driven),而应该是数据驱动(data-driven),即交联剂的优势必须经过实验的充分验证,而不仅仅是设计理念的优势[42]。表1展示了部分交联剂及其特性。一些综述专门针对交联剂的进展进行了总结[42-44],其中德国柏林工业大学Rappsilber 团队[42]的综述详细解剖了交联剂的组成模块,荷兰乌得勒支大学Heck团队[43]的综述详细对比了可断裂交联剂与不可断裂交联剂的异同。
除了使用化学交联剂交联两个氨基酸位点之外,生物体内部也会形成天然的交联形式,例如二硫键交联[45]、类泛素化修饰交联[46]等,此类交联形式称为内源性交联。内源性交联也可通过质谱技术进行鉴定,但鉴定流程的各个环节与图1所示的化学交联质谱技术流程稍有差异。本文将重点介绍化学交联质谱技术,以下简称化学交联质谱技术为交联质谱技术。
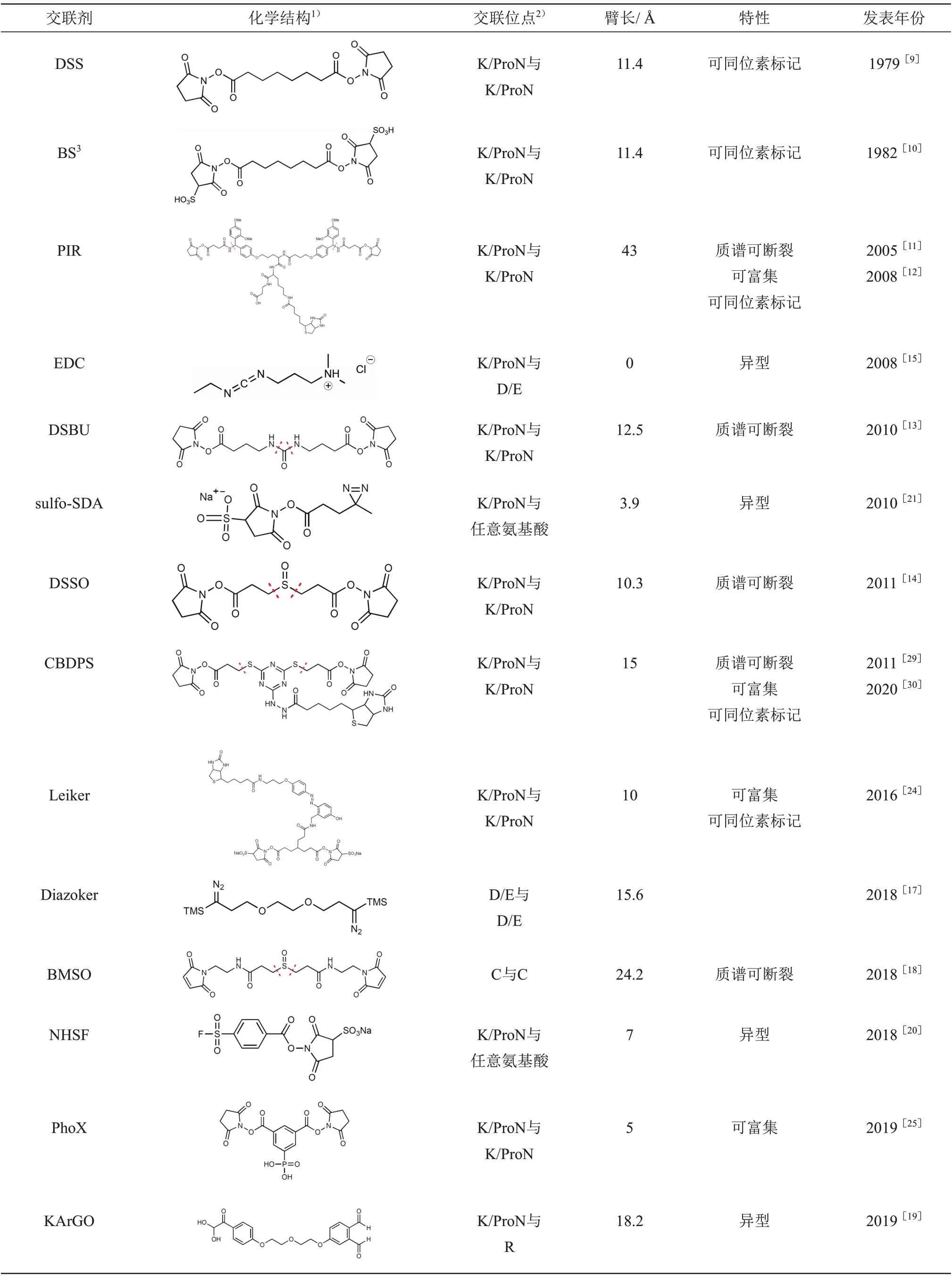
Table 1 The information of some commonly used cross-linkers表1 部分常见交联剂及其特性(以首次发表时间升序排列)
2 交联反应
为了维持蛋白质的活性,交联反应通常需要在合适的条件下进行,需要特别注意的3 个参数是pH、温度和交联剂浓度。过酸、过碱、高温都可能导致蛋白质失活,而且酸性pH还可能导致NHS类交联剂产生较多的副反应[8],所以交联反应通常需要在中性pH 和室温下进行。交联剂的浓度是另一个需要关注的参数,浓度太高不但会导致较多的副反应,还可能影响蛋白质的结构和功能[47]。因此,对于不同的交联剂和蛋白质样品,需要针对性地优化这3个参数。此外,交联反应分为在细胞内(in vivo)进行和细胞外(in vitro)进行,如果想要捕获蛋白质在原生状态下的结构,则需要在细胞内进行交联反应,此时需要选择能够通过细胞膜的交联剂,如DSS[48]和Azide-A-DSBSO[31]等。
3 酶 切
蛋白质发生交联反应之后,需要将交联产物酶切成肽段,以便进入质谱检测。酶切是指蛋白质在酶的催化作用下水解的过程,也称为蛋白质水解。目前常用的酶是胰蛋白酶Trypsin,它能在赖氨酸K和精氨酸R的C端酶切,由于赖氨酸和精氨酸在蛋白质序列中含量较多[49],且它们容易携带正电,使得被Tyrpsin 酶切后的肽段长度适中,且容易被质谱检测。不过,对于交联产物,由于常用的NHS 类交联剂已经交联了赖氨酸,使得被交联的赖氨酸不能被Trypsin酶切,导致Trypsin酶切后的肽段较长、质量较大,可能不易被质谱检测。
后来,有研究人员提出同时使用多种酶进行酶切的并行酶切方法(parallel digestion)[50],即将交联产物分成多份,每份使用不同的酶进行酶切并分别进行质谱数据分析。实验结果表明,并行酶切相比于仅对单份交联产物进行单次Trypsin 酶切能鉴定到更多的交联位点对。然而,有研究结果表明,并行酶切并不会改变酶切肽段的质量分布[51],且如果将交联产物分成多份,每份只使用Trypsin 酶切并分别进行质谱数据分析(相当于多次技术重复),这种只使用Trypsin的并行酶切方法也能达到和使用多种酶的并行酶切方法相当的交联鉴定数目[52]。2019 年,Rappsilber 团队[52]和中国大连化学物理研究所的张丽华团队[53]相继提出使用顺序酶切方法(sequential digestion)对交联产物进行酶切,即先使用Trypsin 酶切,酶切终止之后再加入另一种酶(如Asp-N、Chymotrypsin、Glu-C)进行酶切。顺序酶切能有效降低酶切肽段的长度和质量,使得酶切后产生的交联肽段更容易被质谱检测,进一步也提升了交联位点对的鉴定数目。
交联产物酶切之后,通常会得到4种不同类型的肽段,包括未发生交联反应的线性肽段(linear peptide)、被交联剂一端修饰的线性肽段(monolinked 或type-0 peptide)、肽段内部的交联(looplinked 或type-1 peptide)和肽段间的交联(crosslinked或type-2 peptide pair)。其中,线性肽段由于只含有1条肽段,也称为单肽;肽段间的交联由于含有两条肽段,也称为交联肽段或交联双肽。在这4种类型的肽段中,交联肽段能够提供最多的距离约束信息,是交联质谱技术需要鉴定的主要肽段形式。交联肽段又可以进一步划分为两种:如果交联的两条肽段来自同一条蛋白质,则称为蛋白质内的交联肽段(intra-protein cross-linked peptide pair),否则称为蛋白质间的交联肽段(Inter-protein crosslinked peptide pair)。
4 交联肽段的富集方法
由于交联肽段只占酶切产物的一小部分,大量存在的线性肽段对交联肽段的质谱采集和鉴定带来很大的挑战[6,35]。为了鉴定到更多的交联肽段,需要对其进行富集。现有的交联肽段富集方法大致可分为亲和纯化方法、色谱分离方法和质谱富集方法,不同方法利用了交联肽段的不同特性,下面进行具体介绍。
4.1 亲和纯化
亲和纯化方法利用交联肽段携带交联剂的特性,通过可富集交联剂,实现对交联肽段的富集。可富集交联剂是指交联臂上携带有可富集基团的交联剂。最常见的可富集基团是生物素(biotin),它与链霉亲和素(streptavidin)有很强的亲和作用,因此可以用链霉亲和素磁珠富集交联肽段。生物素的一个不足是体积较大,由于位阻效应(steric effects),可能会影响交联剂的反应效率[4]。常见的携带生物素的可富集交联剂有PIR[11-12]、Leiker[24]、CBDPS[29-30]、PC-DUCCT-biotin[54]等。
为了避免生物素对交联反应效率的影响,生物素的引入可延后到交联反应之后。具体来说,携带炔基(alkyne)或叠氮化合物(azide)的交联剂先进行交联反应,然后通过点击化学(click chemistry)的方式使炔基或叠氮化合物与生物素发生反应,进而使交联剂带上生物素,后续再通过生物素富集交联肽段。由于炔基和叠氮化合物的体积较小,而且几乎不存在于生物分子中,具有较好的生物正交性(biorthogonality),使包含此类基团的交联剂反应效率较高,对生物体影响较小。常见的携带炔基或叠氮化合物的可富集交联剂有Azide-ADSBSO[31]、cliXlink[26]、NNP9[55]等。
2019 年,中国上海科技大学的陈文章团队和Heck 团队相继设计出可富集交联剂pBVS[37]和PhoX[25],作者借鉴磷酸化肽段的富集方法,在交联剂的交联臂上加入磷酸基团,然后分别使用二氧化钛(TiO2) 和固定化金属离子亲和色谱(immobilized metal-ion affinity chromatography,IMAC)富集交联肽段,达到了较好的富集效果。
总的来说,基于亲和纯化的交联肽段富集方法具有很好的设计理念,能高效地富集携带交联剂的肽段,但由于大量被交联剂修饰的线性肽段也携带交联剂,使富集后的交联肽段占比依然不够高[24-25,56],这是此类富集方法普遍存在的一个问题。
4.2 色谱分离
由于交联双肽由两条肽段组成,其电荷和体积往往大于非交联肽段。色谱分离方法即利用交联肽段所带电荷较多、体积较大的特性,通过强阳离子交换(strong cation-exchange,SCX)色谱[57]或分子排阻色谱(size exclusion chromatography,SEC)[50]分离交联肽段和非交联肽段,从而达到富集交联肽段的目的。在强阳离子交换色谱中,固定相中含有带负电的离子交换剂,当流动相经过色谱柱时,带低正电的肽段与固定相的作用力较弱,洗脱较早,而带高正电的肽段与固定相的作用力较强,洗脱较晚,因此交联肽段往往有更长的保留时间。在分子排阻色谱中,固定相中含有大小不同的孔隙,当流动相经过色谱柱时,较小的肽段进入孔隙使得其保留时间较长,而较大的肽段只能从较小的孔隙的外侧绕过,洗脱较早,因此交联肽段往往有更短的保留时间。色谱分离方法对分离交联肽段有一定效果,但如果非交联肽段含有较多的遗漏酶切位点,使得其所带电荷和体积与交联肽段接近时,便难以与交联肽段分离。
4.3 质谱富集
离子淌度分离(ion mobility separation,IMS)是一种在气相中分离复杂离子化混合物的技术,其与质谱技术结合形成的离子淌度质谱(ion mobility mass spectrometry,IMMS)是一种功能强大的分离和分析方法[58]。在离子淌度质谱中,样品被离子化之后,首先进入离子淌度分离设备,该设备充满缓冲气体并被施加了一定强度的电场,不同离子由于质量、电荷、形状等的差异,与缓冲气体碰撞之后通过电场的时间不同;通过改变电场强度,可实现对不同离子的分离;通过离子淌度分离的离子再进入质量分析器进行质谱分析。2020年,德国莱布尼茨分子药理学研究所的刘凡团队[59]使用装备了高场不对称波形离子淌度谱(high field asymmetric waveform ion mobility spectrometry,FAIMS)的Lumos质谱仪,一定程度上分离了交联肽段离子和非交联肽段离子,将HEK293T 样品的DSS交联位点鉴定数目提升了1倍。同年,Heck团队[60]使用装备了捕集离子淌度谱(trapped ion mobility spectrometry,TIMS)的timsTOF Pro 质谱仪,在PhoX 亲和纯化富集的基础上,进一步分离了交联肽段离子和被交联剂修饰的线性肽段离子,有效避免了50%~70%的被交联剂修饰的线性肽段离子被采集二级谱,起到了较好的富集交联肽段离子的效果。
无论是亲和纯化方法,还是色谱分离方法,都是在样品进入质谱前对交联肽段进行富集,即使是离子淌度分离方法,也只是处在色谱和质谱之间的一种分离方法。当样品进入质谱之后,如果能够控制质谱仪,使其采集更多的交联肽段的碎片离子谱图,进而增加交联肽段鉴定数目,从最终效果来看,也相当于“富集”了交联肽段。目前,大多数交联质谱研究工作通常使用数据依赖采集(datadependent acquisition,DDA)方式采集肽段的碎片离子谱图,在这种方式下,质谱仪根据一些规则从一级谱中挑选若干母离子峰,进一步碎裂得到对应的碎片离子谱图。如果能够识别出一级谱中哪些母离子是交联肽段,哪些母离子是非交联肽段,然后控制质谱仪有针对性地采集交联肽段的碎片离子谱图,则能在质谱端实现对交联肽段的富集。2016年,Rappsilber 团队[61]利用交联肽段的母离子质量较大、所带电荷较高的特点,设计了基于决策树的质谱数据采集方案,仅采集电荷超过2+、质量大于1 300 u 的母离子的碎片离子谱图,在这种策略下,能够避免59%的非交联母离子采集二级谱,且只损失2%的交联谱图,一定程度上起到了富集交联谱图的效果。
总的来说,目前的交联肽段富集方法大多是生物化学方法,不同方法利用了交联肽段的不同特征,虽然能起到一定的富集效果,但富集效果还有提升空间。如果能综合不同富集方法提取的特征,使用计算技术训练更加强大的分类器,用于区分交联肽段和非交联肽段,也许能进一步改进富集效果。
5 交联肽段的质谱碎裂方法
液相色谱法-质谱联用(liquid chromatographymass spectrometry,LC-MS,简称液质联用)是指将液相色谱的物理分离能力和质谱的质量分析能力结合起来的分析化学技术。前面4.2已经介绍了使用液相色谱分离交联肽段的方法,下面先简要介绍蛋白质组学中常见的质谱碎裂方法,然后介绍针对交联肽段的质谱碎裂方法。
5.1 常见质谱碎裂方法简介
蛋白质组学中常见的质谱碎裂方法主要有3种,分别是碰撞诱导裂解(collision-induced dissociation,CID)、高能碰撞裂解(higher energy collisional dissociation,HCD) 和电子转移裂解(electron transfer dissociation,ETD)。CID 是利用加速电场,使肽段离子和中性分子发生碰撞,进而碎裂肽段的方法[62]。被CID 碎裂的离子通常在离子阱(ion trap,IT)中扫描成碎片离子谱图,由于母离子碎裂和碎片离子扫描都在离子阱中完成,CID-IT的组合方式具有速度快、灵敏度高的优势。但是离子阱的分辨率和质量精度相对较低,且存在固有的“1/3 效应”,即无法记录质荷比小于1/3 母离子质荷比的碎片离子[63]。后来,有研究人员提出HCD 碎裂方式,即母离子在特定的碰撞室(collision cell)内完成碰撞和碎裂,然后碎片离子被传输到轨道阱(orbitrap,OT)中完成扫描[64]。HCD-OT的组合方式不存在“1/3效应”,具有分辨率和质量精度高、质量范围宽等优势,但采集速度比CID-IT 慢一些。CID 和HCD 主要碎裂肽键,产生b/y离子。
ETD 是一种和CID、HCD 完全不同的碎裂方式,它利用携带电子的阴离子与携带质子的肽段离子之间的反应,使肽段离子发生碎裂[65]。相比于CID 和HCD,ETD 碎裂不受肽段序列的影响,且不易碎裂修饰基团,所以碎片离子包含了完整的修饰质量信息,进而更容易鉴定修饰位点,所以ETD 在整体蛋白质鉴定与修饰鉴定中有广泛的应用[66]。但是ETD 往往存在碎裂不完全的问题,使得碎片离子谱图中存在较强的低价母离子峰。为了提高肽段的碎裂效率,研究人员又提出了ETciD[67]和EThcD[68]的碎裂方式,即在ETD碎裂之后补充CID 或HCD 碎裂。ETD 主要碎裂N-Cα键,产生c/z离子;ETciD和EThcD同时产生b/y和c/z离子。
由于交联剂是否质谱可断裂对交联肽段的质谱碎裂方法有很大影响,下面分别介绍质谱不可断裂与质谱可断裂的交联剂所交联肽段的质谱碎裂方法。为了便于叙述,将质谱不可断裂交联剂所交联的肽段称为不可断裂交联肽段;类似地,将质谱可断裂交联剂所交联的肽段称为可断裂交联肽段。
5.2 不可断裂交联肽段的质谱碎裂方法
由于不可断裂交联肽段的交联剂在质谱中不容易断裂,所以针对此类交联肽段碎裂方法的主要目标是充分碎裂肽段,以利于后续的交联肽段鉴定。2012年,北京生命科学研究所的董梦秋团队[69]利用BS3交联的合成肽段数据集,对比了CID、HCD和ETD的谱图质量,发现HCD 碎裂的交联谱图质量最好(图2a)。因此,对于不可断裂交联肽段,HCD 作为一种简单有效的碎裂方式,被大多数实验室所采用。
后来,Rappsilber 团队[70]进一步在简单的蛋白质复合物样品上对比了CID、HCD、ETD、ETciD 和EthcD 5 种碎裂方案,再一次证明了HCD在碎裂不可断裂交联肽段上的优势。此项工作表明,HCD 在碎裂大多数交联肽段时具有较好的序列覆盖度,且具有速度优势,能鉴定到更多的交联肽段;EThcD 在碎裂高价态、大质量交联肽段时,能获得比HCD 更高的序列覆盖度,但代价是更长的碎裂时间。因此,作者提出了一种基于决策树的碎裂方案,即根据母离子的质荷比与电荷,实时决定采用HCD 或EThcD 碎裂方式。此后,该团队一直使用此决策树方案碎裂交联肽段[71-72]。由于ETD 碎裂功能只存在部分质谱仪中,EThcD 的应用范围受限。
除了HCD 碎裂方法之外,也有研究人员利用紫外光裂解(ultraviolet photodissociation,UVPD)碎裂不可断裂交联肽段。相比于HCD,虽然UVPD碎裂方式鉴定到的交联肽段数目更少,但可作为HCD的补充,能鉴定到一些HCD鉴定不到的交联肽段[73]。
5.3 可断裂交联肽段的质谱碎裂方法
对可断裂交联肽段的碎裂需要同时兼顾两个方面,一方面希望能碎裂交联剂得到两条肽段的完整肽段离子峰,以便推断出交联两条肽段的质量,进而降低鉴定复杂度(见6.2);另一方面,又希望能充分碎裂肽段,得到尽可能多的肽段碎片离子,以便鉴定肽段序列。然而,这两个目标往往是互相矛盾的,因为高丰度的完整肽段离子峰意味着肽段碎裂不充分,所以产生的肽段碎片离子较少。此外,由于交联剂中易断裂键的键能往往小于肽键,使得交联剂比肽段更容易在低能量下碎裂,导致单次碎裂往往无法同时获得足够的完整肽段离子峰和肽段碎片离子峰。

Fig.2 Fragmentation methods for cross-linked peptide pairs图2 交联肽段质谱碎裂方法
为了同时获得足够的完整肽段离子峰和肽段碎片离子峰,研究人员相继提出了多种不同的碎裂方案(图2b~d)。在可断裂交联剂提出之初,研究人员普遍采用三级质谱的碎裂方案[11,13-14,74](图2b):先用低能量CID 碎裂肽段母离子;如果在二级谱中能检测到丰度高、且质量差为某个预设值的双峰(doublet),则说明该母离子是交联肽段,且双峰是已分离的两条单肽的肽段离子峰;接着,隔离双峰离子,进行三级质谱碎裂,得到两条单肽的碎片离子谱图。在这种方案下,交联两条单肽的质量可从二级谱中推算得到,结合三级谱图,可利用常规单肽搜索引擎鉴定交联双肽的序列。三级质谱的碎裂方案由于对交联两条单肽分开进行碎裂和扫描,避免了交联双肽碎裂时互相影响的问题[75],也简化了碎片离子谱图。但是,三级质谱的触发取决于能否在二级谱中找到固定质量差的双峰,如果可断裂交联肽段在二级谱中没有形成双峰,或者质谱仪找到的双峰并不是肽段离子峰,则可能导致漏打或误打三级谱的问题。
2015 年,Heck 团队[76]提出了CID+ETD 的组合碎裂方案(图2c):对于每一个肽段母离子,同时进行低能量CID碎裂和ETD碎裂,CID碎裂提供肽段离子双峰和少量b/y碎片离子,ETD 碎裂提供c/z碎片离子。在这种方案下,交联两条单肽的质量可从CID 二级谱中推算得到,结合CID 和ETD二级谱中的碎片离子峰,可利用常规单肽搜索引擎鉴定交联双肽的序列。进一步,该团队在2017 年将三级质谱方案和CID+ETD 方案组合,提出了CID-MS2-MS3-ETD-MS2 的混合方案[77],即对同一个肽段母离子,同时使用三级质谱方案和CID+ETD 方案碎裂,且新增了基于谱峰强度的三级质谱触发机制。实验结果表明,新的混合方案相比于单独使用三级质谱方案或CID+ETD 方案,能显著提高交联肽段鉴定数目。然而,使用ETD 的方案要求质谱仪支持ETD 碎裂,这一定程度上限制了方案的普适性。此外,上述方案都需要对母离子进行多次碎裂和扫描,数据采集效率较低。
2016 年以来,有多个研究团队相继提出了基于阶梯能量的HCD 碎裂方案(stepped collision energy HCD,SCE-HCD)(图2d),如德国哈雷-维滕贝格马丁路德大学的Sinz 团队[78]、澳大利亚新南威尔士大学的Wilkins团队[79]和奥地利分子病理学研究所的Mechtler 团队[80]。SCE-HCD 方案对每一个肽段母离子,分别使用低、中、高三种能量进行HCD 碎裂,然后把三次碎裂的离子扫描到一张二级谱中。在SCE-HCD碎裂方案中,低能量HCD起到与低能量CID 类似的效果,即主要碎裂交联剂,生成完整肽段离子峰;高能量HCD 主要碎裂肽段主干,生成碎片离子峰。由于质谱仪参数限制,阶梯能量必须设置3种能量,所以一般还会设置1 个折中的中等能量。对于DSBU,由于交联剂断裂的键能与肽段主干断裂的键能相当,针对DSBU 交联肽段的3 个碎裂能量相对集中,在Fusion 质谱仪中一般为27-30-33[78];对于DSSO,由于交联剂断裂的键能比肽段主干断裂的键能更低,针对DSSO 交联肽段的3 个碎裂能量相对分散,在Lumos中一般为21-27-33[80]。对于PIR,虽然暂未有SCE-HCD 碎裂方案发表,但针对PIR 的碎裂方式也已经从三级质谱[74]过渡到仅使用二级质谱[81],可以预见SCE-HCD对PIR也有进一步提升效果。
SCE-HCD 碎裂方案概念简单,数据采集效率较高,且不受仪器的限制。已有的工作表明,无论是DSBU 还是DSSO,SCE-HCD 相比于前几种碎裂方案,都能鉴定到更多的交联肽段[78-80]。虽然SCE-HCD 的优势已初步显现,但因为其将完整肽段离子峰和肽段碎片离子峰扫描在同一张二级谱图中,而可断裂交联双肽的肽段离子峰通常有4 根,相当于4条肽段共碎裂形成了1张混合谱图,对后续的碎片离子谱图解析带来较大的挑战。总的来说,对于可断裂交联肽段,目前存在多种不同的碎裂方案,每种方案都存在一定的不足,SCE-HCD相对来说优势更加明显,有望成为碎裂可断裂交联肽段的通用方法。
6 交联肽段的鉴定方法
相比于常规单肽鉴定,交联肽段鉴定具有更大的挑战,主要体现在以下3个方面[1,82]:
a.产物更加多样。正如第3节所述,交联样品酶切之后存在至少4种不同类型的肽段,而且交联肽段仅占其中很小一部分,产物的多样性给算法和软件架构设计带来更大的挑战。
b.谱图更加复杂。交联肽段的碎片离子谱图中存在多种类型的离子,既包含常规单肽离子,也包含携带交联剂的交联离子,还包含两条肽段同时碎裂形成的内部离子,谱图的复杂性给匹配打分算法带来更大的挑战。
c.搜索空间更大。交联肽段由两条单肽组合而成,因此其搜索空间随数据库规模的增长而呈平方量级的扩大。据统计,对于人类数据库,交联肽段的搜索空间是常规单肽的数百万倍[1],平方搜索空间问题给鉴定算法带来更大的挑战。
在上述3个挑战中,相对来说,平方搜索空间问题带来的挑战最大,特别是从大数据库中鉴定交联肽段时,巨大的搜索空间对速度和精度都有很大的影响[83]。为了缓解平方搜索空间问题,领域发展出两条不同的技术路线:第一条路线是通过计算技术实现大数据库下的交联肽段鉴定;第二条路线是设计可断裂交联剂,通过生化技术避开平方搜索空间问题.下面分别介绍这两条技术路线。
6.1 不可断裂交联肽段的鉴定方法
不可断裂交联剂是使用时间最长、应用范围最广的一类交联剂,自本世纪初交联质谱技术提出以来[2],领域内提出了大量的算法鉴定不可断裂交联肽段[1,82](表2)。在这个过程中,鉴定算法大致经历了4个发展阶段,依次是:基于一级质谱的鉴定算法[2,84]、生成交联肽段数据库借助常规单肽引擎进行鉴定的算法[85-86]、穷举式搜索算法[87-89]和开放式搜索算法[27,69,90]。其中,前两种算法由于没有或只利用了部分交联肽段的碎片离子,鉴定性能较差,现今已经很少被人所使用,下面重点介绍穷举式和开放式搜索算法。
给定一张二级谱图,穷举式搜索算法是指枚举所有的肽段组合,将组合质量加交联剂质量等于母离子质量的交联双肽都与谱图进行匹配打分,取最高分的交联双肽作为该谱图的鉴定结果,如软件StavroX[87]、SIM-XL[91]、Xilmass[88]等都是穷举式搜索引擎。然而,由于平方搜索空间问题,如果数据库中有N条肽段,则朴素版穷举式搜索算法的时间复杂度将为O(N2),难以支持大规模数据库的交联肽段搜索。后续,有研究人员将肽段按质量有序排列,一定程度上加速了穷举搜索的过程[92-93],但肽段排序通常需要O(N*lg(N))的时间,这种方法仍然具有较高的时间复杂度。
2016 年以来,中国香港科技大学的余维川团队持续对朴素的穷举算法进行了改进,先后推出了ECL[89]、ECL2[94]和Xolik[95]三款穷举式搜索引擎。其中,ECL 使用简化的打分算法加速搜索过程; ECL2 使用可加性打分(additive score function)和肽段质量分桶机制(binning strategy)实现了线性时间复杂度O(N)的穷举搜索算法;Xolik 使用双端队列(double-ended queue)和记忆化(memoization)进一步降低了线性时间复杂度的常数系数,可以搜索人类数据库。虽然ECL 和Xolik 的系列工作从理论上加速了交联肽段的穷举搜索过程,但它们在一定程度上牺牲了搜索精度,例如可加性打分无法考虑交联双肽所有可能的碎片离子、对母离子误差精度要求高等。从实际评测结果来看,Xolik 搜索引擎的精度还有较大的提升空间[56]。
2008 年,美国华盛顿大学的Goodlett 团队[90]首次将开放式搜索策略引入到交联肽段鉴定中来,提出了开放式搜索引擎Popitam。此后,开放式搜索策略逐渐成为鉴定交联肽段的主流方法。在这一策略中,交联双肽被看作两条单肽各自携带了一个大质量修饰,修饰质量可通过母离子质量减去单肽质量得到。因此,开放式搜索首先将数据库中的单肽与谱图进行开放式粗打分,然后将粗打分前k名的单肽组合为交联双肽,再与谱图进行细打分。在开放式搜索策略中,粗打分的时间复杂度为O(N),细打分的时间复杂度为O(k2),因此,总的时间复杂度为O(N+k2)。由于k为较小的常数,开放式搜索的时间复杂度远小于穷举式搜索。常见的开放式搜 索 引 擎 有pLink 1[69]、Protein Prospector[75]、Kojak[96]等,它们的主要差异在于k的取值不同,分别为k=500、k=1 000、k=250。值得一提的是,除了k的差别之外,在粗打分保留候选单肽时,不同策略也会导致引擎性能的差异。pLink 1 以母离子质量的一半为界,将候选单肽划分为稍大质量的α肽段和稍小质量的β肽段,然后α肽段和β肽段各自保留前k名;Protein Prospector和Kojak并不区分α肽段和β肽段,而是将所有候选单肽作为一个整体保留前k名。由于大质量肽段的碎片离子偏多,粗打分往往偏高,Protein Prospector 和Kojak 的策略有可能无法在前k名中召回正确的β肽段,导致搜索失败。相对来说,pLink 1将α肽段候选和β肽段候选分开保留的策略更加合理,灵敏度更高。
常规开放式搜索的时间复杂度为O(N+k2),虽然相比于穷举式有很大进步,但当数据库规模N很大时,开放式搜索的粗打分阶段会成为新的性能瓶颈。自2014 年以来,中国科学院计算技术研究所的pFind 团队[97]在pLink 1 工作的基础上,着手研发新一代开放式交联肽段搜索引擎,并最终于2019年推出新版pLink 2[56]。pLink 2利用交联双肽碎裂不均的特点[75],设计了两阶段的搜索策略,即先搜索碎裂较好的偏长肽段,后搜索碎裂一般的偏短肽段。此外,又分别设计了肽段碎片质量索引和完整肽段质量索引加速长肽段和短肽段的搜索过程。实验结果表明,pLink 2 相比于pLink 1 速度提 升 40 倍。 此 后, xiSEARCH[52,61]和MetaMorpheusXL[98]也采用了类似的两步搜索策略或肽段碎片质量索引结构。此外,pLink 2 通过引入新的预处理算法pParse[99]和半监督机器学习重打分算法Percolator[100-101],在精度方面也有所改善,是目前整体性能最佳的交联肽段搜索引擎之一。
6.2 可断裂交联肽段的鉴定方法
可断裂交联剂相比于不可断裂交联剂的优势在于,交联剂断裂可分离交联的两条肽段,并在二级质谱中形成完整肽段离子特征峰,根据特征峰可推算出交联两条单肽的质量,进而将交联肽段鉴定问题简化为两次常规单肽鉴定问题。所以对于可断裂交联肽段的鉴定,核心问题是如何从二级质谱中找出特征峰,并推算出交联两条单肽的质量。
对于常见的可断裂交联剂,如DSSO、DSBU和PIR,其交联臂上有两个断裂位点。如图3所示,当使用低能量CID对交联肽段进行碎裂时,如果交联臂左边的位点断裂,则会生成带交联剂短臂的αS和带交联剂长臂的βL两根特征峰;如果交联臂右边的位点断裂,则会生成带交联剂长臂的αL和带交联剂短臂的βS两根特征峰;如果两个位点同时断裂,则还可能生成报告离子峰r(reporter ion)。对于DSSO 和DSBU,其报告离子质量分别为49.98 u 和25.98 u,由于质量太小,通常无法被二级质谱所检测;对于PIR系列交联剂,其报告离子质量较大(如BDP-NHP 的报告离子质量为751.41 u[81]),通常可以被二级质谱所检测,可用来判断二级谱是否为交联谱图。
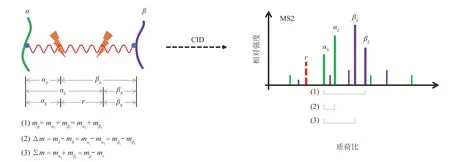
Fig.3 Three methods to derive masses of two peptides linked by cleavable cross-linkers图3 可断裂交联剂简化交联肽段鉴定的3种方法
给定一张可断裂交联肽段的二级谱图,通常有3 种方法从谱图中推算出交联两条单肽的质量(图3):
a.枚举任意两根峰,如果它们的质量之和等于母离子质量,则认为找到αS和βL或αL和βS,使用此类方法的搜索引擎有Link-Finder[14]和MaXLinker[102];
b.枚举任意两根峰,如果它们的质量之差等于交联剂报告离子质量,则认为找到αL和αS或βL和βS,使用此类方法的搜索引擎有XlinkX[76-77]和MeroX[78,103];
c.枚举任意两根峰,如果它们的质量之和等于母离子质量减报告离子质量,则认为找到了αS和βS,使用此类方法的搜索引擎有ReACT[74]和Mango[81]。
其中方法a 和方法c 利用了母离子质量,方法b 仅利用了碎片谱峰的质量;3 种方法都同时适用于DSSO、DSBU 和PIR 交联肽段。最近刚发表的MS Annika则同时使用了上述多种方法[104]。
由于并不是所有可断裂交联肽段都能在二级谱中形成完整的4 根特征峰,如果缺失特征峰较多,上述基于特征峰质量的单肽质量推算方法将失效。为了提升灵敏度,有些搜索引擎利用特征峰强度较高的特点,增加了基于特征峰强度的单肽质量推算方法。如XlinkX 2.0假设强度排名前三的谱峰中存在1根特征峰,则可通过母离子质量推算出另一条肽段的特征峰质量,进而推算出两条单肽的质量[77]。当得到两条单肽的质量之后,可以使用常规单肽搜索引擎进一步鉴定两条单肽的序列,如Mango 使 用Comet[105]、 MS Annika 使 用MS Amanda[106]等。
需要指出的是,无论是三级质谱(图2b)还是CID+ETD(图2c)的碎裂方式,可断裂交联肽段都是在低能量CID碎裂下形成特征峰,在这种碎裂方式下,二级谱中的肽段离子特征峰的强度较高、碎片离子相对较少。因此,无论是基于特征峰质量还是特征峰强度,都能相对容易推算出两条单肽的质量。但是,如果是在高能量HCD或者SCEHCD(图2d)的碎裂方式下,则二级谱中会混杂大量碎片离子谱峰,而且携带交联剂的碎片离子也满足上述一些质量关系或者谱峰强度较高,给单肽质量推算带来较大的挑战。
最后,如果不考虑可断裂交联肽段的碎裂特性,则现有的不可断裂交联肽段搜索引擎也可用于鉴定可断裂交联肽段。不过大多数不可断裂交联肽段搜索引擎由于在匹配打分时没有考虑交联剂和肽段主干同时断裂产生的碎片离子,它们的灵敏度还有待提升。此外,由于没有利用肽段离子特征峰的信息,不可断裂交联肽段搜索引擎在鉴定可断裂交联肽段时的搜索空间依然是平方量级,搜索速度也还有待提升。表2展示了部分常见交联肽段搜索引擎的信息。
Table 2 The information of some commonly used search engines for cross-linked peptide pairs表2 部分常见交联肽段搜索引擎(以首次发表时间升序排列)
7 交联肽段鉴定的质量控制方法
基于数据库搜索的交联肽段鉴定方法,一般都会发生随机匹配的情况,使得鉴定结果集合中存在错误的结果。如何估计鉴定结果集合中的假发现率(false discovery rate,FDR)是质量控制需要解决的核心问题。2012 年之前,有研究工作使用打分经验阈值[33]、随机交联剂质量[85]、晶体结构比对[27]等方法估计FDR,但这些方法判定标准不一致、主观性大、不易推广。2012年,pFind团队[69]和瑞士分子系统生物学研究所的Aebersold团队[113]同时提出了使用目标诱饵库方法(target-decoy approach,TDA)估计交联FDR的方法。该方法凭借简单有效、容易推广的优点,逐渐成为估计交联FDR的一般性方法,一直沿用至今。2012年以来,TDA 估计交联FDR 的方法有了进一步发展,同时也出现了其他估计交联FDR 的方法,下面分别进行介绍。
7.1 基于目标诱饵库的假发现率估计方法
TDA 方法是指在数据库搜索时,除了搜索目标数据库,还搜索一个与目标库特征相似的诱饵库,然后利用鉴定结果中匹配到诱饵库的比例,估计匹配到目标库的结果中随机匹配错误结果的比例。在错误结果随机匹配到目标库和诱饵库的概率相等的前提下,可推导出交联肽段鉴定的FDR 估计公式为:
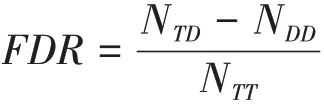
其中,NTT和NDD分别表示两条肽段均匹配到目标库或诱饵库的谱图数目,NTD表示一条肽段匹配到目标库、另一条肽段匹配到诱饵库的谱图数目。
随着对TDA 方法研究的深入,后续有多项工作对该方法进行了补充和完善。TDA 方法提出之初,对于是否该将蛋白质间交联和蛋白质内交联结果分开估计FDR,领域内存在不同的做法[69,113]。2014 年,美国加州大学旧金山分校的Chalkley 团队[75]分析发现,如果将两者合并估计FDR,则会低估蛋白质间交联的FDR、高估蛋白质内交联的FDR,因此建议分开估计两者的FDR,简称为分开过滤策略。2015年,pLink-SS[107]和Kojak[96]分别在鉴定二硫键交联肽段和化学交联肽段时,都使用了分开过滤的策略。2019 年,pFind 团队[56]通过理论推导和实验验证,论证了分开过滤策略的合理性和有效性。
2017年,Rappsilber团队[114]的研究工作表明,如果仅在交联谱图层次控制FDR,然后将交联谱图结果归并到交联位点或蛋白质相互作用层次时,由于正确结果相对聚集、错误结果相对分散,高层次的FDR 会急剧上升,因此,应该直接在交联位点或蛋白质相互作用层次控制FDR。最近,Rappsilber 团队[115]推出新版xiFDR,可估计蛋白质相互作用层次的FDR,并尝试用实验方法检验其FDR估计的可靠性。
7.2 合成肽段检验方法
合成肽段检验是指利用合成肽段交联数据集检验引擎性能的方法,该方法通常和陷阱数据库检验方法相结合,共同检验搜索引擎的性能。合成肽段检验的基本思想是,利用合成肽段交联数据集构建一个高可信的标注集,然后让搜索引擎搜索标注集,使用的数据库中除了包含合成肽段序列,还额外添加一个与合成肽段无关的数据库(称为陷阱库)作为干扰;如果搜索引擎的鉴定结果与标注结果一致,则认为鉴定正确,否则认为鉴定错误。下面介绍两个合成肽段检验的代表性成果。
2012 年,董梦秋团队和pFind 团队[69]合成了38 条肽段,两两交联之后共获得741 个交联数据集,然后利用人工标注的方法构建了一个包含2 077 张谱图的标注集,此标注集是迄今为止规模最大的合成肽段交联标注集。该团队利用此标注集搜索陷阱库的方法,先后检验了pLink 1[69]和pLink 2[56]两个引擎的性能。2015年,同一团队使用类似的方法,构建了一个包含2 289张谱图的合成肽段二硫键交联标注集,利用该标注集检验了pLink-SS搜索引擎的性能[107]。
2020年,Mechtler团队[116]设计了另一种合成肽段检验方法,该团队将95 条合成肽段划分为12组,每组有7~9条肽段;组内肽段交联之后再将12组交联产物混合在一起进行质谱分析。Mechtler团队的合成肽段交联实验理论上可产生434组交联双肽,数据规模少于董梦秋团队和pFind团队的合成肽段交联实验;而且由于组内肽段的数目较多,且将多组交联产物混合进行质谱分析,前者的实验设计不如后者精细。但是,Mechtler团队使用该数据集设计了一种自动化的结果检验方法。在Mechtler团队的实验设计下,组间肽段的交联候选相当于陷阱库,如果搜索引擎鉴定到组间肽段的交联结果,则认为错误;反之,如果鉴定到了组内肽段的交联结果,则认为正确。合成肽段检验可以较为客观地评估鉴定结果的错误率,是评估搜索引擎性能的理想方法。然而合成肽段检验的成本较高,相关数据集规模较小,难以大范围推广应用。
2021 年,Rappsilber 团队[115]将Mechtler 团队的设计思路拓展到E.coli组学样品,该团队利用分子排阻色谱将E.coli样品分馏为44 份,每份样品内部发生交联反应,然后将44 份交联产物混合在一起进行质谱分析。类似地,如果搜索引擎鉴定到馏份内部的蛋白质交联,则认为正确,反之则认为错误。该方法相比于Mechtler团队的方法,无需合成肽段,可在蛋白质相互作用层次检验复杂样品的交联鉴定结果。但是,由于样品变得更加复杂之后,同一馏份内部的交联搜索空间增大,使得馏份内部交联的随机匹配概率增大,进而影响该检验方法的可靠性。
7.3 其他方法
一直以来,大多数工作使用蛋白质晶体结构检验交联位点鉴定结果的正误[27,69,75,88,96],如果交联位点之间的距离小于晶体结构中记录的距离,则认为鉴定正确,否则认为鉴定错误。近年来,有多项工作对晶体结构检验方法提出了质疑。一方面,蛋白质在溶液中存在动态变化的构象,和晶体结构中记录的信息不完全一致,即使不满足晶体结构中的距离约束,也不能完全说明鉴定结果错误[117-118]。另一方面,并不是所有蛋白质都存在晶体结构信息,如果仅估计存在晶体结构信息的交联位点子集的错误率,且用该错误率代表鉴定到的交联位点全集的错误率,则可能极大低估全集的错误率[119]。
pFind团队提出了15N代谢标记检验的质量控制方法,并成功应用于常规单肽[120]、糖肽[121]和交联双肽[56]的结果检验中。该方法是一种干湿结合的质量控制方法,以交联双肽结果检验为例,其样品制备过程如下:首先,分别以无标记培养基和15N 标记培养基培养大肠杆菌E.coli细胞,则后者的蛋白质中所有的N元素都被代谢标记为重标形式;然后,无标记样品和15N 标记样品分别进行交联反应;最后,将无标记交联产物和15N 标记交联产物以1∶1 比例混合,并进行质谱数据采集。在这种情况下,每一对交联双肽的母离子在一级谱中都存在轻、重两簇同位素峰,且谱峰强度的比值接近1∶1。因此,15N代谢标记的检验过程为:首先,搜索引擎搜索无标记交联双肽;然后,计算鉴定到的交联双肽的无标记和15N 标记的母离子质荷比;最后,在一级谱中,计算无标记和15N 标记的母离子峰的强度比值,如果比值接近1∶1,则认为鉴定结果正确,否则认为鉴定结果错误。15N 代谢标记检验方法概念简单,不需要合成肽段,可大规模批量检验鉴定结果。不足之处是仅仅依靠一级谱图无法识别N 元素与正确肽段相等的错误鉴定结果,且无法标记人类等细胞。
除了晶体结构检验和15N 代谢标记检验,还存在其他多种检验方法,例如陷阱数据库检验[56,102-103,119]、 蛋 白 质 相 互 作 用 数 据 库 检验[102-103,119]、正交方法检验[102,119]、多种碎裂谱图检验[122]等。所有这些检验方法,都有共同的特点。2019 年,pFind 团队[123]的研究工作表明,任何一种检验方法都存在将正确结果误判为错误(错报)或将错误结果误判为正确(漏报)的情况,因此可以统一使用错报率和漏报率两个指标评价各种质量控制方法。例如,基于TDA 的FDR 估计方法相当于将诱饵库作为陷阱库的陷阱数据库检验方法,由于诱饵库中的蛋白质序列并不存在于样品中,因此鉴定到诱饵库的结果一定为错误,其错报率约为0;而鉴定到目标库的结果不一定完全正确,在错误结果随机匹配到目标库与诱饵库的概率相等的情况下,其漏报率约为50%。利用类似的方法,该团队还分析了15N 代谢标记检验的错报率与漏报率[56]。质量控制方法的蓬勃发展,说明领域对鉴定结果的准确度的关注度越来越高,预计交联肽段的精准鉴定将成为新的研究热点。
8 交联质谱技术的应用
交联质谱技术主要用于辅助解析蛋白质结构和研究蛋白质相互作用。由于交联剂臂长的限制,交联反应只能发生在空间距离足够接近的两个氨基酸之间,如果鉴定到的两个交联位点来自同一条蛋白质(蛋白质内交联),则说明蛋白质的三维结构中两个位点之间的距离不超过交联剂的臂长,因此交联距离约束可辅助解析蛋白质结构;如果鉴定到的两个交联位点来自不同的蛋白质(蛋白质间交联),则说明两条蛋白质的交联位点区域距离接近,可能存在相互作用关系,因此交联距离信息可用于研究蛋白质相互作用。
8.1 蛋白质结构解析
解析蛋白质的结构对了解蛋白质的功能具有重要作用。交联质谱技术凭借对蛋白质纯度要求低、样品量要求少的优点,成为解析蛋白质结构的很好的互补技术[3,5-6,49]。例如交联质谱技术和X 射线晶体衍射技术(X-ray crystallography)结合,解析了Prp19 同源四聚体[124]和泛素结合酶UBE2S[125]等的结构;和核磁共振技术(nuclear magnetic resonance,NMR) 结合,解析了人类Hsp90/FKBP51 蛋白质混合物[126]和纤维母细胞生长因子FGF21[127]等的结构。2012年,得益于软硬件技术的突破,冷冻电镜技术(cryo-electron microscopy,Cryo-EM)成为解析蛋白质结构的重要方法之一[128]。不过,冷冻电镜对蛋白质的柔性区域(flexible domains)不能提供足够的结构信息,而交联质谱技术正好可以弥补这一不足。因此,交联质谱技术和冷冻电镜技术相结合成为蛋白质结构解析非常流行的方法[129]。自2013年以来,交联质谱技术和冷冻电镜技术结合解析了大量蛋白质复合物的结构,例如酵母剪接体[130]、人源mTOR2 复合体[131]和转录共激活复合物SAGA[132]等,体现了交联质谱技术的独特优势。
8.2 蛋白质相互作用研究
蛋白质通过与不同蛋白质或核酸分子相互作用形成精细、动态的网络来调节各项生命过程。交联质谱技术通过交联反应将两个氨基酸共价连接,能较好地固定微弱的、瞬时的相互作用,在研究蛋白质相互作用上具有独特优势[133]。例如,使用交联质谱技术研究细菌ADP-Hep 蛋白质与宿主ALPK1激酶的相互作用[134]和酵母转录终止因子Seb1 与RNA聚合酶Pol II 的相互作用[135]等。此外,交联质谱技术还可以与免疫共沉淀 (coimmunoprecipitation, Co-IP)[136]、 酵 母 双 杂 交(yeast two-hybrid,Y2H)[137]和 亲 和 纯 化 质 谱(affinity purification-mass spectrometry, APMS)[138]等技术结合,共同研究蛋白质相互作用。
8.3 蛋白质组水平的交联鉴定里程碑
近年来,得益于交联剂(第1 节)、酶切技术(第3 节)、富集技术(第4 节)、搜索引擎(第6节)等的全面发展,全蛋白质组水平的交联质谱研究越来越多,且不断刷新交联鉴定纪录。2008年,Aebersold 团队[27]利用同位素标记交联剂DSS-D0/D12 和搜索引擎xQuest 在E. coli中鉴定到71 对交联位点。2012 年,董梦秋团队联合pFind 团队[69]利用无标记交联剂BS3和搜索引擎pLink 在E. coli和C. elegans中分别鉴定到394 和39 对交联位点;2016年,该团队利用可富集交联剂Leiker将E.coli和C.elegans的交联位点鉴定纪录提升了一个数量级,分别达到3 130 和893 对交联位点[24]。2013年,美国华盛顿大学的Bruce 团队[74]利用可断裂交联剂PIR 和搜索引擎ReACT 在E. coli中鉴定到708 对交联位点。2015 年,Heck 团队[76]利用可断裂交联剂DSSO 和搜索引擎XlinkX,在HeLa 上鉴定到2 179对交联位点,引起了领域对可断裂交联剂的广泛关注;2017 年,该团队利用优化的质谱和鉴定流程,将HeLa 的交联位点鉴定纪录提升到3 301对交联位点[77]。此后,研究人员利用多种交联剂[79,139-140]、多种酶切[52-53]、充分分馏[102,140-141]等方法,不断刷新全蛋白质组水平的交联鉴定纪录。2020 年,Rappsilber 团队[140]同时使用不可断裂交联剂DSS和可断裂交联剂DSSO,以及搜索引擎 xiSEARCH, 在 肺 炎 支 原 体 细 胞 (M.pneumoniae)中鉴定到12 509对交联位点,是目前交联鉴定的最新纪录。表3列出了目前全蛋白质组水平的交联质谱研究工作。
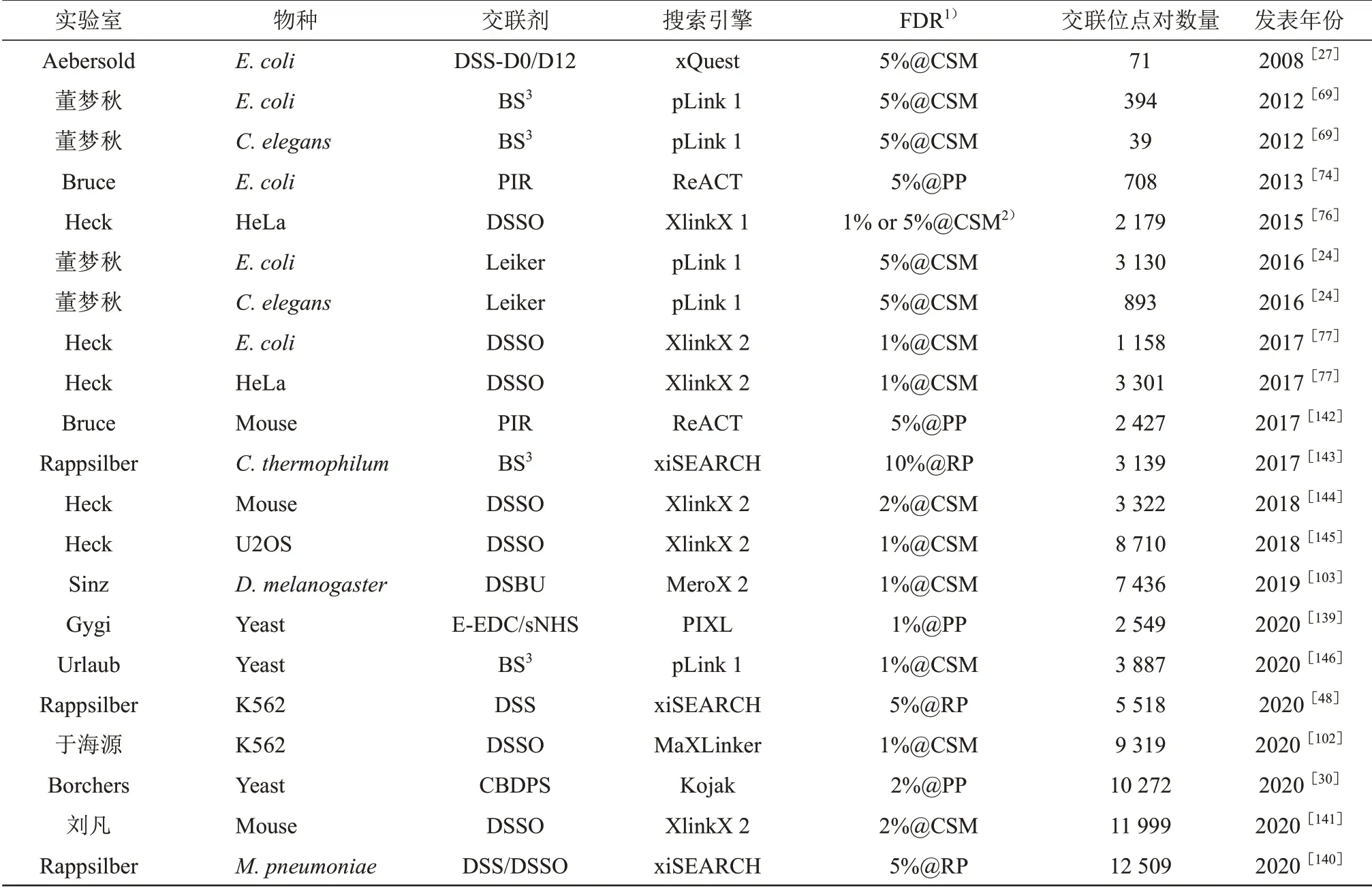
Table 3 The information of some proteome-wide CXMS studies表3 部分全蛋白质组水平的交联质谱研究工作
9 总结与展望
蛋白质结构和相互作用研究对了解蛋白质的功能具有重要作用,然而由于这一研究的复杂性,单一技术往往难以取得很好的效果,生物学家往往会综合多种技术共同研究蛋白质结构和相互作用。交联质谱技术能够提供氨基酸位点之间的距离信息,并且具有通量高、对蛋白质的纯度要求低、可固定弱相互作用等优势。经过二十多年的发展,交联质谱技术的各个环节都取得了长足的进展,已成为整合结构生物学(integrative structural biology)的重要工具之一[3,147]。
交联质谱技术虽然在方法和应用上都取得了很大的进步,但在精准鉴定、深度解析和深度覆盖3个方面还有很大的提升空间。精准鉴定方面,虽然交联FDR 估计公式的提出为交联肽段鉴定的质量控制做出了巨大的贡献,但现有的质量控制方法还可以进一步完善及充实。一方面,虽然领域开始意识到更高层次FDR 控制的必要性[114,148],但目前大多数交联肽段搜索引擎仅支持谱图或肽段层次的FDR 估计,如何估计并验证更高层次的FDR,目前还没有定论。另一方面,基于TDA 的FDR 估计方法本质是利用诱饵库作为陷阱库的质量控制方法,每一种质量控制方法都难以全面刻画鉴定结果的好坏,如何设计互补甚至正交的质量控制方法是值得研究的课题之一。
深度解析方面,目前的交联质谱数据的解析率普遍偏低,不足50%[56]。相对而言,常规单肽质谱数据的解析率已经达到70%~85%,基本实现了质谱数据的深度解析[120]。单肽质谱数据的深度解析主要得益于开放式搜索引擎的推出,如MSFragger[149]、Open-pFind[120]等,它们在鉴定单肽时扩大了搜索空间,考虑了意外酶切、意外修饰等情况,使得解析率大幅提升。交联质谱数据的深度解析既面临与单肽质谱数据的深度解析同样的问题,如意外酶切、意外修饰等,又存在其独有的难点,如交联位点多[48,52]、交联产物多[150]等。此外,交联肽段鉴定本身存在平方搜索空间的问题,如果进一步扩大搜索空间,不但可能严重增加搜索时间,而且可能导致解析率不增反降[83]。因此,如何在扩大交联搜索空间的同时,既能切实地提升交联质谱数据的解析率,又能控制搜索时间在合理范围内,是交联质谱数据深度解析面临的巨大挑战。
深度覆盖方面,虽然目前的交联位点鉴定数目已经突破一万(表3),但Rappsilber团队[5]保守估计人类全蛋白质组交联实验中至少存在20 万对交联位点,如何鉴定到更多的交联位点是深度覆盖面临的核心问题。上文提到的交联质谱数据的深度解析是尽可能多地解析二级谱图,然而交联质谱数据中存在大量的单肽谱图,即使实现了深度解析,对交联位点深度覆盖的提升作用也有限。鉴定交联谱图是鉴定交联位点的基础,为了实现交联位点的深度覆盖,需要采集并鉴定更多的交联谱图,其中采集更多的交联谱图属于深度覆盖的工作。为此,首先需要开发更多的半特异或非特异交联剂,从源头上产生更多的交联组合[23,139];其次需要设计更加高效的交联肽段富集方法,使得进入质谱仪的交联肽段比例更高[24,59-60];最后需要改进质谱仪的母离子选择算法,例如识别一级谱中的交联母离子,只对交联母离子采集二级谱图[61]。因此,交联位点的深度覆盖需要湿实验团队和干实验团队通力合作,湿实验团队需要开发更加高效的交联剂、交联肽段富集方法和质谱数据采集方法,干实验团队需要开发更加高效的母离子选择算法和交联肽段鉴定算法。
