过渡金属元素Sc、Cr和Mn对Mg2Ge掺杂的第一性原理研究
2022-04-18戴松利梁永超马家君
戴松利 梁永超 马家君
(贵州大学大数据与信息工程学院,新型光电子材料与技术研究所,贵阳 550025)
0 引言
Mg2Ge作为一种半导体材料,无毒且制作成本低[1],因其优越的性能(如高热稳定性、大泽贝克系数、低电阻率、低热导率[2⁃4])在光电器件及热电器件中有潜在的应用前景。掺杂是调控半导体电子结构以及理化性质的重要手段。因此,为扩大Mg2Ge在微电子领域的应用范围,在实验和理论上对Mg2Ge的掺杂研究成为了近年来备受瞩目的研究热点。
Liu等[5]在Mg2Ge作为锂离子电池负极材料的研究中发现,随着Li的嵌入量增大,材料实现了由半导体性到金属性再到半金属性的转变。Bai等[6]通过第一性原理计算研究了Bi元素掺杂对Mg2X(X=Si、Ge、Sn)力学性能的影响,发现Bi元素的掺杂能使Mg2X由脆性材料转变为塑性材料。Gao等[7]采用钽管熔接热压法制备了掺Sb的Mg2Ge化合物,相比本征Mg2Ge有更高的电导率和功率因子。Chuang等[8]通过磁控溅射生长Ag掺杂的Mg2Ge薄膜,有效增大了薄膜的晶粒尺寸和表面粗糙度。Yücel[9]通过Be、Ca、Co、Cr、Cu、Fe、Mn、Pd和Zn对Mg2Ge进行掺杂的第一性原理计算研究,对本征Mg2Ge的光学和热电性质进行了有效的调控。
研究人员在实验及理论方面对Mg2Ge进行了大量的掺杂研究,证明了Mg2Ge是一种有广泛应用前景的半导体材料,但对扩大Mg2Ge在自旋电子器件及稀磁二极管的应用的相关研究还未见报道。通常半导体掺杂过渡元素以及稀土元素后,可将其制备成为半金属磁体和稀磁半导体,它们可以利用电子的电荷运动和电子自旋产生磁矩[10]。比如:Kebabi等[11]分别研究了Cu、Ag和Au对单壁氮化硼纳米管的磁学性能的影响,其中Cu和Ag原子掺杂后的氮化硼纳米管被成功诱导磁化产生较大的磁矩;Wang等[12]使用过渡金属(Cr、Mn、Fe和 Co)对 TiO2进行掺杂,其中Cr和Mn掺杂体系表现为半金属磁体,Fe和Co掺杂体系表现为金属磁体;Wu等[13]发现MgO纳米片能够通过过渡金属元素掺杂在费米能级附近产生一定杂质态,从而减少了带隙,成为了比本征半导体带隙更窄的稀磁半导体。因此,基于密度泛函理论(DFT),我们采用过渡金属元素Sc、Cr和Mn对Mg2Ge进行掺杂的第一性原理计算研究,旨在对其电学、磁学以及光学性质的调控提供理论依据。
1 结构模型与计算方法
1.1 结构模型
Mg2Ge晶体是一种反萤石结构,属于正常价金属间化合物,其空间点群为Fm3m,Bravais晶格为面心立方结构,晶格常数a=0.639 nm。如图1a所示,Mg2Ge晶体结构由8个Mg原子和4个Ge原子构成,其中Mg原子位于晶体中8个小立方体的体心位置,Ge原子位于晶体的8个顶角及6个面心位置。在本次计算中,采用1×1×1的超胞(Mg8Ge4)。如图1b所示,分别采用过渡元素Sc、Cr和Mn原子对Mg2Ge晶胞中位于(0.75,0.75,0.75)的Mg原子进行替位式掺杂,掺杂率为8.3%,属于重掺杂。
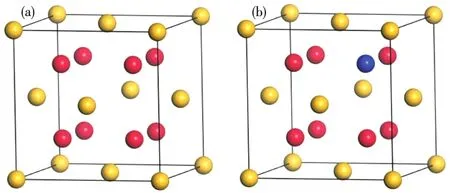
图1 Mg2Ge晶体结构:(a)未掺杂;(b)掺杂后Fig.1 Crystal structure of Mg2Ge:(a)undoped system;(b)impurity doped system
1.2 计算方法
采用基于DFT[14]的CASTEP(Cambridge serial energy package)模块进行计算。为了使模拟的体系达到稳定状态,首先采用BFGS(Broyden⁃Fletcher Goldfarb⁃Shanno)算法对设置极化条件下的Mg2Ge晶胞及其掺杂结构进行结构优化,再对体系的性质进行计算。计算过程选择超软(ulrtrasoft)赝势平面波,交换关联能使用广义梯度近似(generalized gradient approximation,GGA)中的PBE(Perdew⁃Bruke⁃Ernzerhof)方法。为了保证足够的精度,采用7×7×7的Monkhorst⁃Park形式的高对称K点处理布里渊区的积分,平面波截断能设置为450 eV,迭代过程中收敛精度(SCF)设置为每原子5.0×10−7eV,作用在每个原子上的力(Max.force)不大于0.3 eV·nm−1,晶体内应力(Max.stress)不大于0.05 Gpa。参与计算原子的价电子:Mg为 2p63s2;Ge为 4s24p2;Sc为 3d14s2;Cr为3d54s1;Mn为3d54s2。
2 结果与讨论
2.1 几何结构及其稳定性
表1为Mg2Ge晶胞掺杂前后的晶格常数、总能以及掺杂形成能。掺杂形成能Ef[15⁃16]是表征原子掺杂难易程度和掺杂体系稳定性的物理量,掺杂形成能越低,说明掺杂越容易,结构稳定性越高。Ef的定义是:
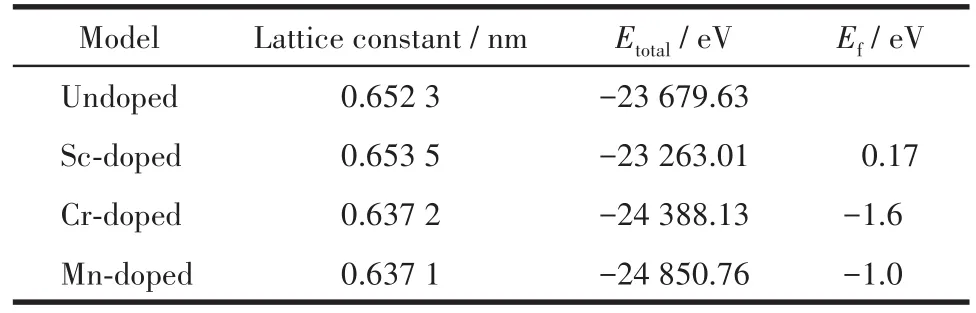
表1 掺杂前后Mg2Ge晶胞的晶格常数、总能和掺杂形成能Table 1 Lattice constant,total energy,and doping formation energy of Mg2Ge cell before and after doping
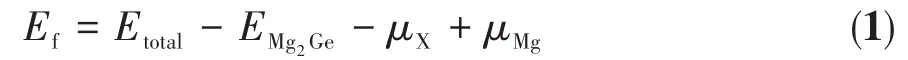
其中Etotal指掺杂后的Mg2Ge晶胞的总能,EMg2Ge为本征Mg2Ge晶胞的总能,μX和μMg分别表示掺杂原子和Mg原子的化学势,其值为对应单质晶胞在充分结构弛豫后得到的平均原子能量。
由表1可知,随着Sc、Cr和Mn原子的掺入,与本征结构相比,Sc掺杂体系的晶格常数增大,Cr和Mn掺杂体系的晶格常数减小,掺杂前后晶格常数变化趋势与以往的工作一致[9]。从原子半径的角度分析,杂质原子与被替换的原子存在原子半径差异,在结构优化过程中,晶胞中的原子由于正负电荷位置重新调整而使库仑相互作用变化,晶格因而发生膨胀或者缩小。Sc的原子半径(0.162 nm)大于Mg的原子半径(0.160 nm),晶格常数增大;Cr的原子半径(0.128 nm)和Mn的原子半径(0.127 nm)小于Mg的原子半径(0.160 nm),晶格常数减小。
比较各掺杂体系的掺杂形成能,Sc掺杂的Mg2Ge掺杂形成能最大且为正值,表明该掺杂体系吸收了能量,由此可知Sc掺杂导致本征Mg2Ge结构稳定性降低。Cr和Mn掺杂体系的掺杂形成能为负值,说明体系放出了热量,因此Cr和Mn掺杂体系增强了本征Mg2Ge的结构稳定性。
2.2 能带结构
由于本次计算是考虑自旋极化条件下进行的,因此能带被分为2部分:自旋向上和自旋向下的能带结构。由图2可知,自旋向上和自旋向下的能带结构完全对称,说明本征Mg2Ge不具有磁性。导带底和价带顶均位于G点,因此本征Mg2Ge为直接带隙半导体,这与Yücel[9]的计算结果一致。禁带宽度Eg=0.25 eV,低于实验值 0.60~0.73 eV[17⁃18],这是由于PBE泛函往往会低估带隙宽度,此处带隙不影响后续物理机制分析。
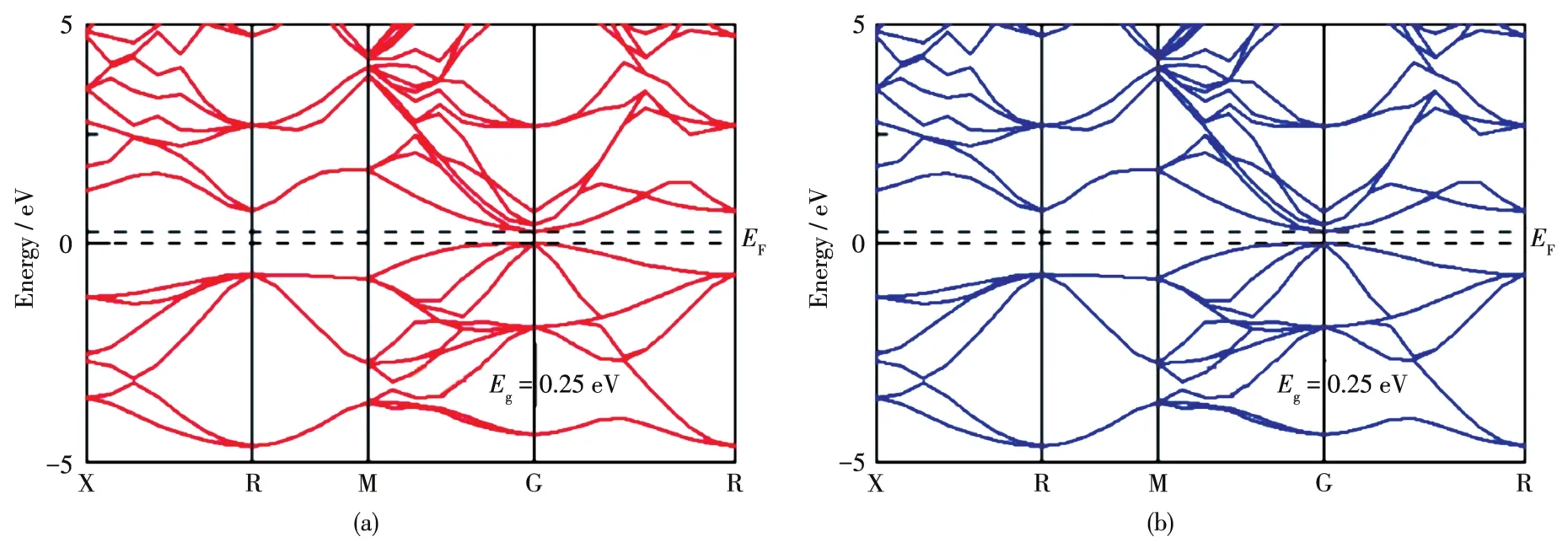
图2 掺杂前的Mg2Ge能带结构:(a)自旋向上和(b)自旋向下Fig.2 Band structure of undoped Mg2Ge:(a)spin⁃up and(b)spin⁃down
图3为Sc掺杂的Mg2Ge的能带结构,自旋向上和自旋向下的能带结构完全对称,表明Sc掺杂体系为非磁性材料。与未掺杂时相比,掺杂后有更密集的能级。由于Sc替代Mg的掺杂属于非等电荷掺杂,Sc原子与周围的Ge原子和Mg原子形成共价配位之后,多余的电子会参与导电,使体系中的电子浓度大于空穴浓度,大量电子从禁带迁移至导带,费米能级从禁带进入导带,体系导电性大大增强,故Sc掺杂后的Mg2Ge为n型简并半导体。
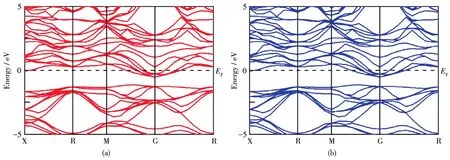
图3 Sc掺杂Mg2Ge的能带结构:(a)自旋向上和(b)自旋向下Fig.3 Band structure of Sc⁃doped Mg2Ge:(a)spin⁃up and(b)spin⁃down
图4为Cr掺杂Mg2Ge的能带结构。在费米能级附近,自旋向上和自旋向下轨道能级发生了明显的劈裂,这表明Cr掺杂体系产生了磁性。自旋向上中费米能级贯穿于杂质带中,自旋向下中费米能级未占据费米面,表现为自旋注入,即杂质带中通过有效质量传输转化为100%的自旋极化载荷子,因此该体系表现为半金属性。
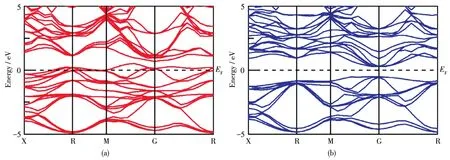
图4 Cr掺杂Mg2Ge的能带结构:(a)自旋向上和(b)自旋向下Fig.4 Band structure of Cr⁃doped Mg2Ge:(a)spin⁃up and(b)spin⁃down
图5为Mn掺杂Mg2Ge的能带结构。在费米能级附近,自旋向上和自旋向下轨道能级发生了明显的劈裂,这表明Mn掺杂体系产生了磁性。费米能级靠近自旋向上的价带顶部,靠近自旋向下的导带底部,呈半导体性。因此,Mn掺杂体系为稀磁半导体。
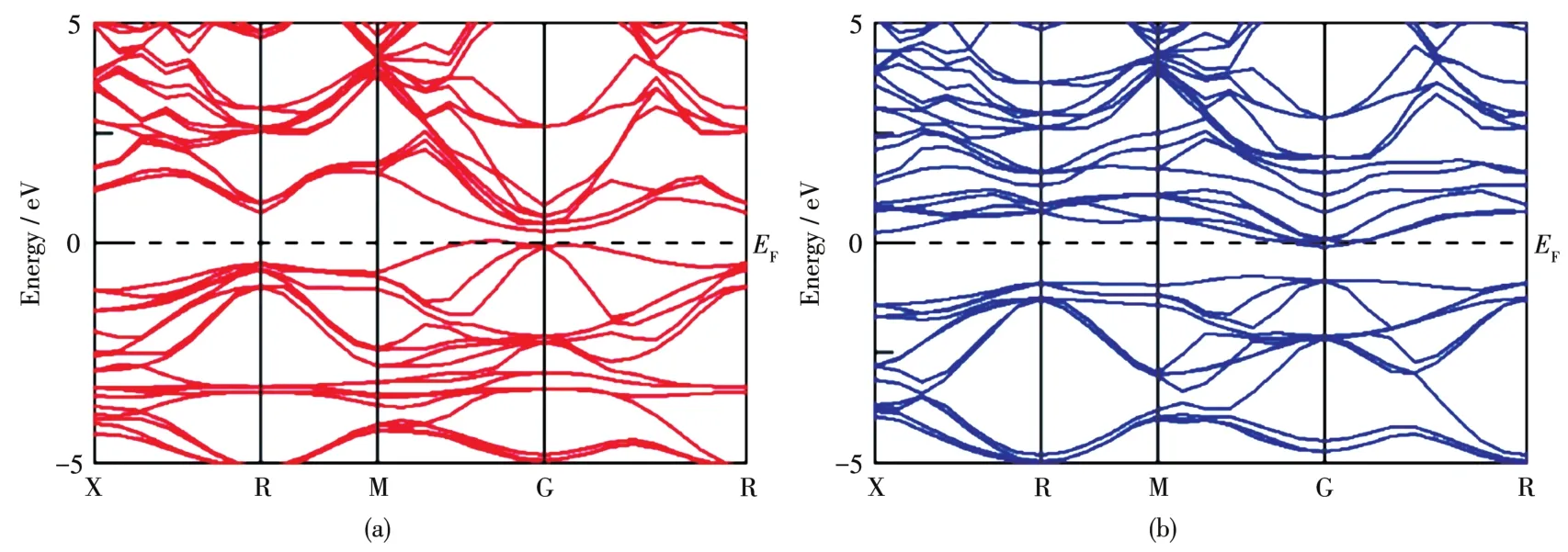
图5 Mn掺杂Mg2Ge的能带结构:(a)自旋向上和(b)自旋向下Fig.5 Band structure of Mn⁃doped Mg2Ge:(a)spin⁃up and(b)spin⁃down
2.3 态密度
为进一步研究掺杂对Mg2Ge电子结构的影响,计算了Mg2Ge掺杂前后的总态密度(TDOS)及各分波态密度(PDOS)。图6为掺杂前后的Mg2Ge的态密度图。由图6a可知,−10~−5 eV的态密度主要由Mg3s态、Mg2p态和Ge4s态电子共同贡献;−5~0 eV的态密度主要由Mg3s态、Mg2p态和Ge4p态共同贡献;0~15 eV区间,Mg2p态为TDOS提供主要的贡献,Mg2s态、Ge4s态和Ge4p态提供少量贡献。费米能级附近的能带组成:价带部分主要由Mg2p态和Ge2p态杂化构成;导带主要由Mg2s态、Mg2p态和Ge4s态杂化构成。自旋向上和自旋向下的TDOS完全对称,说明本征Mg2Ge为非磁性材料。
图6b为Sc掺杂Mg2Ge的态密度图。与本征Mg2Ge不同,它的费米能级附近的TDOS整体向低能区移动,费米能级穿过导带,表现为n型简并半导体特性。价带部分主要由Mg2p态、Ge4s态和Sc3d态杂化构成,其中Sc3d态贡献最少;导带部分主要由Sc3d态、Mg3s态、Mg2p态和Ge4p态杂化构成,其中Sc3d态贡献最大。由此可知,Sc3d态导致大量电子跃迁至导带,电子填充的能量逐渐升高,最终费米能级穿过导带。同时,自旋向上态和自旋向下态完全对称,体系不具备磁性。
图6c为Cr掺杂Mg2Ge的态密度图。掺入Cr原子后,TDOS在费米能级附近发生了自旋劈裂,体系产生了净磁矩。费米能级穿过了自旋向上的价带中,位于自旋向下的带隙中,体系呈半金属性。Mg3s态、Mg2p态、Ge4s态、Ge4p态和Cr3d态的自旋向上和自旋向下态电子数均不相同,在Mg2p态、Ge4p态和Cr3d态中两者差别较为明显,因此体系磁矩主要由Cr3d态、Mg2p态和Ge4p态贡献,其中Cr3d态的贡献最大。在费米能级附近,Cr3d态分别与Mg3s态、Mg2p态、Ge4s态和Ge4p态产生了共振峰,表明在费米能级附近发生p⁃d和p⁃s杂化作用。显然,Cr3d态的自旋向上密度与自旋向下密度不对称,诱导极化了费米能级的Mg2p态和Ge4p态电子产生自旋,使费米能级附近的TDOS发生偏移,体系产生磁矩。对费米能级以下的占据态进行积分计算,体系净磁矩为3.999 9μB,体系中Mg原子平均产生的磁矩为0.03μB,Ge原子平均产生的磁矩为−0.18μB,Cr原子产生的磁矩为4.5μB,体系磁矩分布与态密度分析结果一致。
图6d为Mn掺杂Mg2Ge的态密度图。费米能级附近的TDOS发生了自旋劈裂,表明体系产生了净磁矩。Mg3s态、Mg2p态、Ge4s态、Ge4p态和Mn3d态的自旋向上和自旋向下态电子数均不相同,在Mg2p态、Ge4p态和Mn3d态中两者差别较为明显,因此体系磁矩主要由Mn3d态、Mg2p态和Ge4p态贡献,其中Mn3d的贡献最大。在费米能级附近,Mn3d态分别与Mg3s态、Mg2p态、Ge4s态和Ge4p态产生了共振峰,表明在费米能级附近发生p⁃d和p⁃s杂化作用。显然,与Cr掺杂体系同理,Mn3d态的自旋向上密度与自旋向下密度不对称,诱导极化了费米能级的Mg2p态和Ge4p态电子产生自旋,使费米能级附近的TDOS发生偏移,体系产生磁矩。相比Cr掺杂体系,Mn掺杂体系态密度整体向低能区移动,各个电子轨道在费米能级以下填充电子数增大,因此磁矩较大。经计算体系净磁矩为4.924 6μB,Mg原子平均产生的磁矩为0.041μB,Ge原子平均产生的磁矩为0.1μB,Mn原子产生的磁矩为4.26μB,体系磁矩分布与态密度分析结果一致。
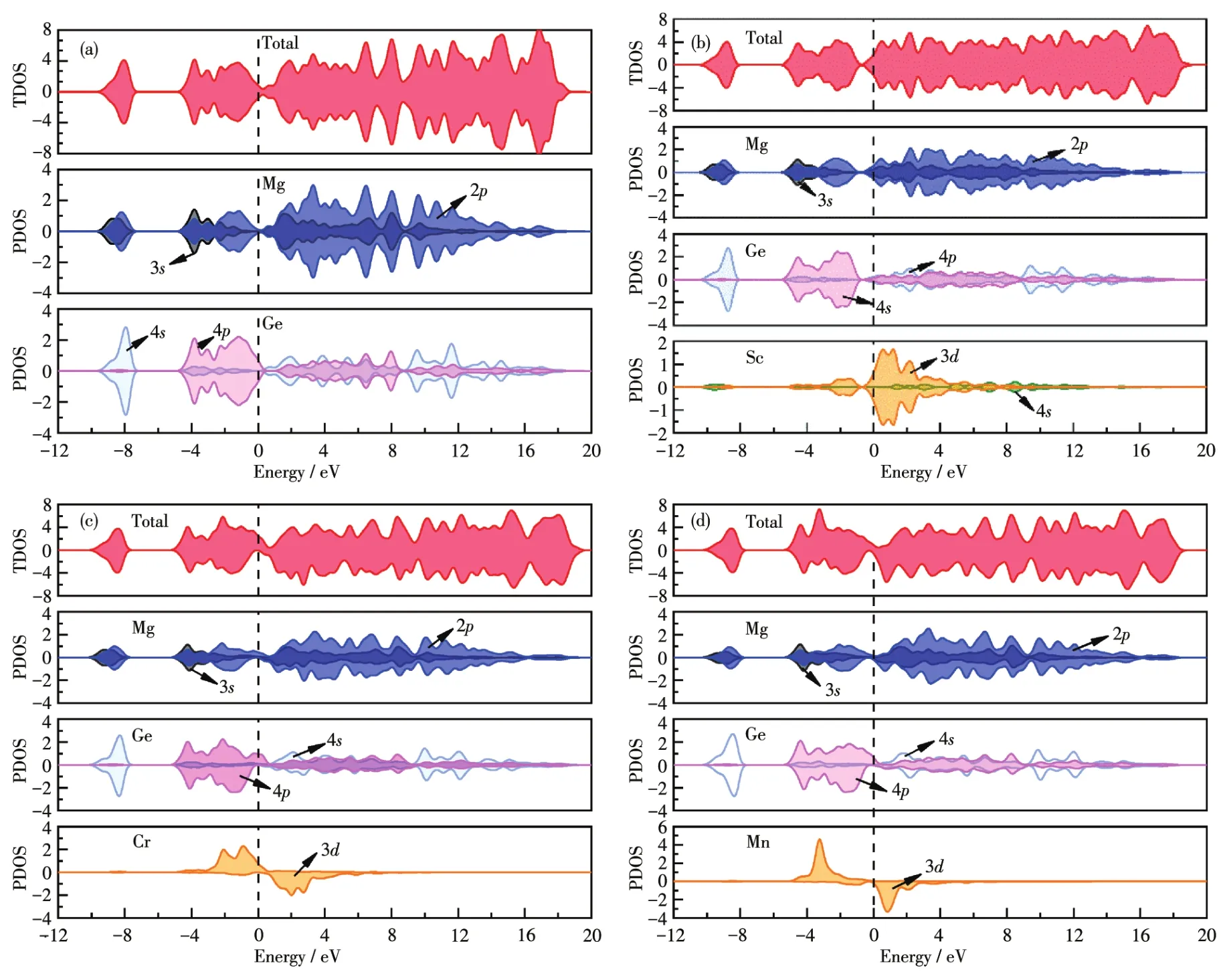
图6 TDOS和PDOS:(a)Mg2Ge;(b)Sc掺杂的Mg2Ge;(c)Cr掺杂的Mg2Ge;(d)Mn掺杂的Mg2GeFig.6 TDOS and PDOS:(a)Mg2Ge;(b)Sc⁃doped Mg2Ge;(c)Cr⁃doped Mg2Ge;(d)Mn⁃doped Mg2Ge
2.4 光学性质
为了研究Sc、Cr和Mn掺杂Mg2Ge的光学性质,计算了掺杂前后Mg2Ge的介电函数与光吸收谱,并分析其影响机制。
介电函数可用于描述系统对电磁辐射的线性响应,并决定电磁波在介质中的传播行为,是沟通微观物理粒子跃迁过程与固体电子结构关系的桥梁,且能反映固体能带结构。通过介电函数可以得到各种光谱信息。介电函数的表达式如下:

其中ε1(ω)是介电函数的实部,ε2(ω)是介电函数的虚部,二者可以由折射率n(ω)和消光系数k(ω)得出:

ε1(ω)可以根据直接跃迁概率定义以及Kramers⁃Kronig色散关系[19]求出;ε2(ω)可以由占据态和非占据态波函数矩阵元素得到。所有的光学性质,如吸收系数等均可以由ε1(ω)和ε2(ω)推导而出,本文计算有关的内容如下[23]:
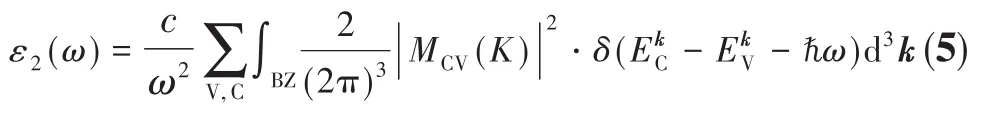
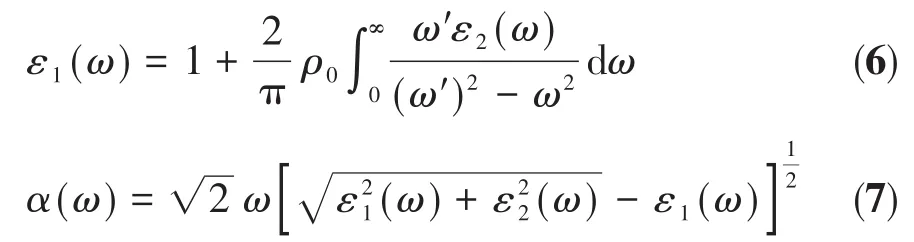
式5中BZ表示第一布里渊区,C和V分别表示导带和价带,c为光速,δ代表狄拉克δ函数,ℏ为约化普朗克常数,k为倒格矢,ω和ω′分别为末状态和初状态的角频率,ρ0为极化响应,|MCV(K)|2为动量跃迁矩阵元,和分别为导带和价带上的本征能级[20],α代表吸收系数。以上关系是分析固体能带结构分布和吸收光谱分布的理论根据。
图7为掺杂前后Mg2Ge的介电函数的实部和虚部。当光子能量为零时,介电函数实部对应数值为静态介电常数,由图7a可知,未掺杂的Mg2Ge的静态介电常数为22.84,经Sc、Cr和Mn掺杂后的Mg2Ge的静态介电常数分别为26.85、28.54和25.56,掺杂后静态介电常数得到提升。半导体材料的静态介电常数与禁带宽度关系[21]为ε(0)≈1+(hω′/Eg)2,其中ω′为等离子频率,Eg为半导体的禁带宽度。掺杂后的Mg2Ge的静态介电常数呈这样的关系:εMn(0)<εSc(0)<εCr(0),其禁带宽度Eg,Mn>Eg,Sc>Eg,Cr,与能带计算结果吻合。图7b为掺杂前后Mg2Ge的介电函数虚部,本征Mg2Ge的峰值出现在2.53 eV,Sc、Cr和Mn掺杂后的Mg2Ge峰值分别出现在2.31、2.49和2.36 eV,即发生了红移,且峰值均下降,分析该现象是由多体效应造成的,它导致填充态到非填充态的跃迁能量减小。
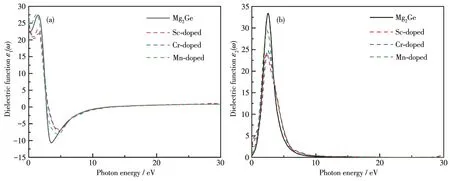
图7 Mg2Ge掺杂前后的介电函数的(a)实部和(b)虚部Fig.7 (a)Real parts and(b)imaginary parts of the dielectric functions for Mg2Ge before and after doping
图8为掺杂前后Mg2Ge的吸收光谱图。由图8a可知,掺杂前后的吸收谱在3.26~12.4 eV的紫外光波段有强烈的吸收峰,即掺杂后总体仍保持了本征结构在该波段的高吸收性。掺杂Sc、Cr和Mn后的Mg2Ge在29.75、42.58和46.88 eV处产生了新的次强峰,吸收光谱都有所扩展,增强了Mg2Ge的光催化性能。掺杂前后Mg2Ge吸收带边均为0.01 eV,说明掺杂不会改变它们的光吸收带边缘。由图8b可知,在红外光波段Mn掺杂体系的吸收系数始终大于本征体系;在近红外光波段的起始阶段,Sc和Mn掺杂体系的吸收系数略小于本征体系,但在波长大于975 nm时,吸收系数逐渐大于本征体系。因此,掺杂体系对近红外光波段的吸收能力得到了提升。
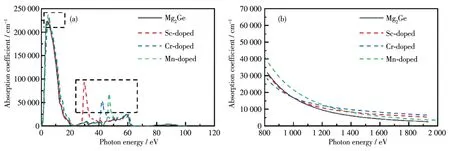
图8 Mg2Ge掺杂前后的(a)吸收光谱图和(b)红外光段范围内的吸收光谱图Fig.8 (a)Absorption spectra and(b)absorption spectra in the infrared spectrum segment for Mg2Ge before and after doping
3 结论
采用基于密度泛函理论的第一性原理赝势平面波方法,计算了过渡金属元素Sc、Cr和Mn掺杂Mg2Ge前后的能带结构、态密度和光学性质,计算结果表明:
(1)未掺杂的Mg2Ge是一种直接带隙半导体,带隙为0.252 eV。Sc掺杂后,费米能级穿过导带,呈n型简并半导体,体系导电性能增强。Cr和Mn掺杂后,Mg2Ge转变为半金属磁体和稀磁半导体,体系净磁矩均由杂质原子3d态电子以及其诱导极化的Mg2p态、Ge2p态自旋电子贡献,整体产生了较为可观的磁矩,为Mg2Ge在自旋电子器件的实际应用提供了理论基础。
(2)经Sc、Cr和Mn掺杂后,Mg2Ge的静态介电常数得到提升,提高了对光的利用率;介电函数虚部主峰发生红移,表明填充态到非填充态的跃迁能量减小;吸收光谱得到了扩展,增强了本征Mg2Ge的光催化活性,保持了紫外光波段的强吸收特性,对近红外光波段的吸收能力得到了提升。
