多重耐药大肠杆菌中前噬菌体的分布特征及诱导分离
2022-04-14刘教刘畅陈进王勉之熊文广曾振灵
刘教,刘畅,陈进,王勉之,熊文广,曾振灵
多重耐药大肠杆菌中前噬菌体的分布特征及诱导分离
刘教,刘畅,陈进,王勉之,熊文广,曾振灵
华南农业大学/广东省兽药研制与安全评价重点实验室/国家兽医微生物耐药性风险评估实验室,广州 510642
【】通过调查前噬菌体在多重耐药大肠杆菌中的分布特征、诱导分离以及前噬菌体中耐药基因与毒力基因的流行状况,为研究前噬菌体介导耐药基因在细菌的传播提供科学依据。挑选前期保存的2018—2019年广东省分离的131株禽源多重耐药大肠杆菌进行核酸提取及全基因组测序,将二代测序的结果组装拼接成全基因组序列,上传至噬菌体PHASTER网络数据库与数据库中已有的噬菌体基因组序列进行比对分析。利用CGE数据库比对耐药基因与毒力基因,从而获得在前噬菌体上耐药基因与毒力基因的分布情况。温和性噬菌体由丝裂霉素C诱导并使用双层平板法分离纯化。131株大肠杆菌药物敏感性试验的结果显示,氨苄西林、四环素、氟苯尼考、复方新诺明的耐药率均高达90%以上,其次是头孢类抗生素以及庆大霉素、环丙沙星、美罗培南和黏菌素均在50%左右,替加环素的耐药率达到了0.2%,所有菌株都呈现出多重耐药的现象,均为多重耐药大肠杆菌。131株多重耐药大肠杆菌中共检出736个前噬菌体片段,其中包含329个完整型前噬菌体,其与40个已知数据库噬菌体物种以不同百分比匹配上;可疑型噬菌体有66个,其与20个已知数据库噬菌体物种以不同百分比匹配上;不完整型噬菌体有341个,其与52个已知数据库噬菌体物种以不同百分比匹配上,完整型前噬菌体的基因序列显示出与已知的噬菌体物种的序列相似性最高,平均为58.53%;131株大肠杆菌中平均前噬菌体数量为5.6个,平均总含量为152.4 kb。前噬菌体基因组占其宿主基因组的比例分布在0.58%—5.87%,以3.0%为主。前噬菌体基因组长度范围在2.8—107.9 kb,其中13.0 kb的前噬菌体出现的频次最高,占所有前噬菌体的9.1%。CGE比对结果表明,131株多重耐药大肠杆菌的基因组共在18株前噬菌体序列检测到耐药基因(A)、(G)和,其中(A)、(G)和检出数分别为16、1和1。71株多重耐药大肠杆菌前噬菌体中携带有6种不同的毒力基因,其中存在部分菌株携带2种或者3种毒力基因,有62株前噬菌体携带端粒酶RNA基因,16株前噬菌体携带血清存活率增加基因,外膜蛋白酶、黏附素基因、和ABC转运蛋白基因分别在2、2、1和1株前噬菌体中检出。(A)和分别是前噬菌体中最常见的耐药基因和毒力基因。温和性噬菌体诱导试验结果显示,前噬菌体的诱导成功率为84.0%,但出现噬菌斑的概率仍比较低。前噬菌体在多重耐药大肠杆菌中分布广泛且携带有多种耐药基因和毒力基因,温和性噬菌体诱导成功率高,具有携带耐药基因及毒力基因水平传播的风险,需要加强和持续监测。
前噬菌体;大肠杆菌;诱导;分布特征;耐药基因
0 引言
【研究意义】噬菌体是感染细菌、真菌、藻类、放线菌或螺旋体等微生物的病毒的总称,因部分能引起宿主菌的裂解,故称为噬菌体。噬菌体可分为溶原性噬菌体和裂解性噬菌体[1]。溶原性噬菌体,也称为温和性噬菌体,由细菌中的前噬菌体诱导产生,主要是通过将自身基因组整合入细菌基因组的方式进行传代。这种带有噬菌体基因组的细菌称为溶原性细菌,而整合入细菌基因组的噬菌体称为前噬菌体[2-3]。温和性噬菌体对其细菌宿主有重要的生态和进化影响,在面对复杂的环境中能够与细菌进行共进化,同时,温和性噬菌体会携带一定的耐药基因,可导致细菌耐药性的传播[4-6]。研究前噬菌体在多重耐药大肠杆菌的分布特性对于细菌耐药性的防控具有重要意义,为深入研究噬菌体通过转导方式介导耐药基因水平基因转移提供基础科学依据。【前人研究进展】耐药基因水平转移(horizontal gene transfer,HGT)是细菌获得耐药基因的重要方式之一,且细菌获得外源性基因的主要方式有接合转移、转化及转导[7-10]。前噬菌体在细菌的基因组上广泛存在,例如在177株鲍曼不动杆菌中鉴定出1 156个前噬菌体,并携带耐药基因包括 OXA-23和NDM-1,前噬菌体是细菌间耐药基因水平转移的重要载体[11-16]。然而,噬菌体介导耐药基因的水平转移起先被认为发生的概率较低,对耐药基因的传播能力比较低,没有引起足够的重视,后逐渐有文章报道其介导的耐药基因水平转移能力不可忽视[17-25],并且已在不同种属细菌中证实,例如四环素和链霉素抗性葡萄球菌[26-27],四环素和氯霉素抗性沙门氏菌鼠伤寒沙门氏菌DT104[5],四环素和庆大霉素抗性肠球菌[28]和耐红霉素的艰难梭菌[16]。【本研究切入点】目前国内外有关噬菌体的研究大多针对于噬菌体通过转导方式介导耐药基因水平转移的机制及噬菌体治疗和展示方向,对于多重耐药大肠杆菌中前噬菌体的流行状况等相关的报道还比较少。而前噬菌体上耐药基因和毒力基因的存在对噬菌体和细菌的共同进化机制有重大影响[29-30]。【拟解决关键问题】本文以多重耐药大肠杆菌中前噬菌体为研究对象,调查前噬菌体在多重耐药大肠杆菌中的分布状况,以及前噬菌体中耐药基因与毒力基因在的流行情况,同时利用丝裂霉素C诱导分离温和性噬菌体,为研究前噬菌体介导耐药基因在细菌的传播提供科学依据。
1 材料与方法
1.1 培养基
LB琼脂培养基(Luria-Bertani Agar),LB肉汤(Luria-Bertani Broth)均购于山东海博生物有限公司;麦康凯琼脂培养基、水解酪蛋白琼脂(Mueller-Hinton Agar)购自广东环凯微生物科技有限公司;Agar购于西格玛奥德里奇(上海)贸易有限公司。
1.2 主要药品试剂
丝裂霉素C购自美国MCE公司;细菌全基因组试剂盒购于广州碧尧德生物科技有限公司;氨苄西林、庆大霉素、替加环素、环丙沙星、美罗培南、黏菌素等12种抗生素均购自生工生物工程(上海)股份有限公司。
1.3 细菌复苏
将甘油肉汤保存的广东省2018—2019年禽源大肠杆菌菌株(来自广东省兽药研制与安全评价重点实验室)划线于麦康凯琼脂培养基上,37℃恒温培养箱培养18—24h。
1.4 药物敏感性测定
使用琼脂稀释法测定细菌的最小抑菌浓度,选用12种针对革兰氏阴性菌的抗生素进行药物敏感性的测定。根据美国临床实验室标准化委员会(CLSI)规定的细菌药敏试验标准,以MIC值位于其规定的敏感、中介、耐药的折点值范围作为判断结果。
1.5 全基因组提取
纯化已复活的菌株,涂于LB琼脂培养基上,过夜培养,备用。按照全基因组测序试剂盒说明书进行提取,将菌液转移至1.5 mL离心管中,离心弃上清,加入适量Buffer STE, Lysozyme和RNase Solution,涡旋混匀,37℃水浴40 min,加250 μL Buffer DL和10 μL Proteinase K至细胞重悬液中。70℃消化10 min,加250 μL无水乙醇,涡旋后转移至吸附柱,10 000×离心1 min,弃去滤液加500 μL Buffer GW1, 10 000×离心1 min,弃滤液加入650 μL Buffer GW2,10 000×离心1 min,倒弃流出液,10 000×空离2 min,柱子装入新1.5 mL离心管,加入50 μL预热至70℃ddH2O,放置3 min,离心弃柱子,-20℃暂放,将样品送至北京诺禾致源生物有限公司进行测序。
1.6 前噬菌体的鉴定与分析
使用CLC genomics workbench version 10.0.1进行组装,通过在线网站(https://cge.cbs.dtu.dk/)进行多基因座序列分型(Multi-locus sequence typing,MLST)。同时,使用PHASTER工具(http://phaster.ca/)进行细菌基因组中前噬菌体的鉴定[31],通过上传基因组与已知的NCBI病毒基因组数据库,转运RNA和噬菌体在细菌基因组上的附着位点进行匹配,然后对检测出来的前噬菌体进行评分,依据分数(总分150)分为完整型噬菌体(Intact,>90)可疑型噬菌体(Questionable,70—90之间)和不完整型噬菌体(Incomplete,<70)。基于前面鉴定的前噬菌体,计算其基因组长度,分析前噬菌体在细菌基因组上的分布特性,并且计算前噬菌体在细菌基因组中的占比情况。同时,将提取到的前噬菌体序列与已知数据库噬菌体物种进行比对,对它们的相似度进行比较分析。
将提取的前噬菌体序列上传至基因组流行病学中心主页网站(https://cge.cbs.dtu.dk//services)进行比对,检测前噬菌体序列上的耐药基因和毒力基因,并对检测到的耐药基因和毒力基因的分布进行分析。
1.7 前噬菌体的诱导
复活前面所用菌株,划线于麦康凯琼脂平板上,于37℃恒温培养箱过夜培养,挑取单菌落接种到已高压的2 mL LB肉汤试管中,37℃,180 r/min摇床培养4 h,用移液枪抽取50 μL菌液至新的含5 mL LB肉汤试管中,摇床培养至OD600为0.3左右,加入丝裂霉素C,使其终浓度为1 μg·mL-1,并设置对照组,继续同样条件摇床培养12 h,测定其OD600值,若OD值下降明显,则认为其诱导成功。
1.8 温和性噬菌体的分离与纯化
划线培养大肠杆菌工程菌C600,接种于5 mL LB肉汤试管中,180 r/min,37℃摇床培养,备用;倒薄层的LB琼脂板备用;将5 mL半固体与100 μL的C600菌液混合加入薄层LB琼脂板中,摇晃混匀,待琼脂凝固后,取诱导液滴至平板中,37℃恒温培养箱培养过夜,次日,观察是否有噬菌斑的产生。若产生噬菌斑,则将诱导液稀释并与菌液1﹕1混合铺双层平板,过夜培养,若有单个噬菌斑,则用枪头扣取噬菌斑,充分涡旋,使噬菌体释放,6 000 r/min,4℃离心5 min,用注射器吸取上清液,过0.22 μm的滤膜,以除去细菌。用双层平板法重复三次完成噬菌斑的纯化[32-33]。
2 结果
2.1 药物敏感性结果
药敏试验结果表明,131株大肠杆菌对试验所用的12种抗生素(氨苄西林、头孢噻肟、头孢他啶、美罗培南、庆大霉素、阿米卡星、四环素、替加环素、氟苯尼考、黏菌素、环丙沙星、复方新诺明)的耐药率分别是96.06%、78.74%、57.48%、44.88%、62.20%、15.75%、93.70%、0.2%、96.06%、53.54%、85.04%和93.70%,呈现出多重耐药现象(图1)。
2.2 多重耐药大肠杆菌中前噬菌体的分布特性
131株多重耐药大肠杆菌中三类前噬菌体(完整型,可疑型和不完整型)基因组分析结果(图2)显示,前噬菌体占其宿主基因组的比例为0.58%—5.87%,大部分在3%左右,平均每株多重耐药大肠杆菌中前噬菌体数量为5.6个,其平均总含量为152.4 kb。统计736个前噬菌体的不同基因组长度的概率分布结果发现前噬菌体基因组大小显示出一个清晰的峰值,峰值噬菌体大小为13 kb,占所有736株前噬菌体的9.1%,前噬菌体最大长度可达到107.9 kb,占比为0.14%,最小的基因长度为0.28 kb,占总前噬菌体的0.27%(图3)。

AMP,氨苄西林;CTX,头孢噻肟;CAZ,头孢他啶;MEM,美罗培南;GEN,庆大霉素;AMI,阿米卡星;TET,四环素;TIG,替加环素;FLR,氟苯尼考;CL,黏菌素;CIP,环丙沙星;SXT,复方新诺明
2.3 大肠杆菌中前噬菌体的鉴定与相似度比较
131株多重耐药大肠杆菌的基因组中累计检测到736个潜在的前噬菌体序列片段,其中包含了329个完整型前噬菌体(Intact prophage),66个可疑型前噬菌体(Questionable prophage)和341个不完整型前噬菌体(Incomplete prophage);其分别与40、22和52个已知数据库噬菌体物种以8.33%—95%不同百分比匹配上。
由图4可知,完整型前噬菌体的基因序列显示出与已知的噬菌体物种的序列相似性最高,平均为58.53%,剩下的两种前噬菌体(可疑型噬菌体和不完整噬菌体)与已知的噬菌体序列相似度比较低,其平均相似度分别是46.58%和37.83%,其原因可能是完整的前噬菌体的序列更长,或者序列中匹配源ORF数量多导致与不完整噬菌体序列相比有更高的物种序列相似度。另外,不同的ST型都与特定的已知噬菌体匹配,其匹配的与已知的噬菌体相似率也极其相似。mEp460样噬菌体、Fels_2样噬菌体和SfⅡ样噬菌体在完整前噬菌体序列上分布最广且相似率最高(图5)。mEp460样噬菌体在ST162、ST167、ST48、ST5229和ST6725上均有分布;SfⅤ样噬菌体主要是分布在ST10中;Fels_2样噬菌体主要分布在ST162、ST48、ST746中;SfⅡ样噬菌体则主要是分布在ST10、ST746、ST23中;仅在ST10中检测到P1样噬菌体、HK75样噬菌体,仅在ST7085中检测到Psp3样噬菌体(图5)。
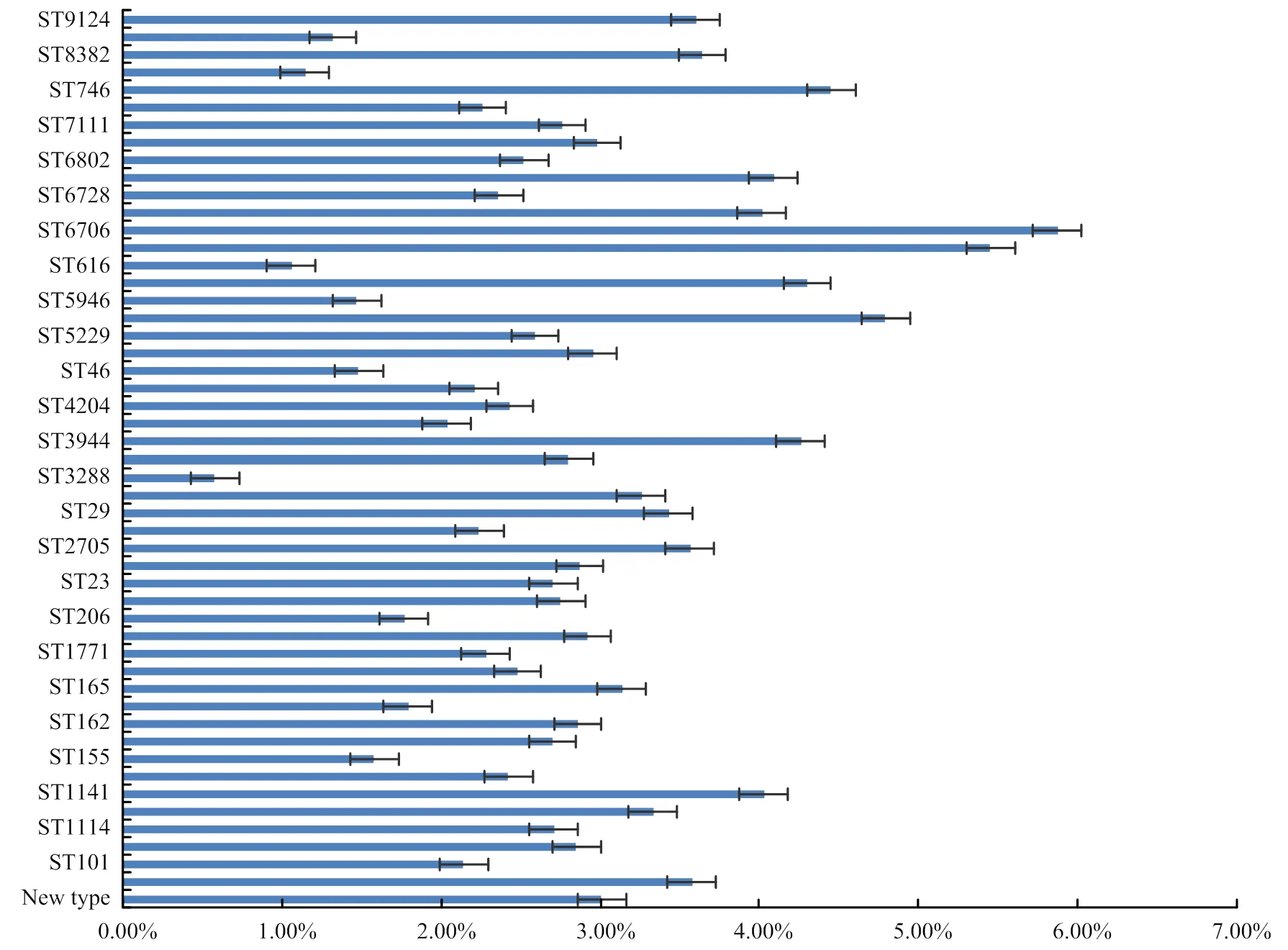
图2 前噬菌体占其宿主基因组的长度贡献百分比

图3 前噬菌体不同基因组长度概率分布图
2.4 耐药基因和毒力基因在前噬菌体序列上的分布特性
耐药基因与毒力基因检出结果(图6)表明,131株多重耐药大肠杆菌的基因组共在18株前噬菌体序列检测到耐药基因(A)、(G)和,其中(A)、(G)和检出数分别为16、1和1,其中16个耐药基因在完整型前噬菌体上分布,2个在不完整型前噬菌体上分布,可疑型前噬菌体上未找到耐药基因。(A)基因可作为多药耐药的转运体,携带(A)基因的菌株对氯霉素、四环素和红霉素等抗生素均有耐药性[34]。(G)属于林可霉素类耐药基因在不完整前噬菌体上发现,该前噬菌体与PHAGE__SJ46相似度最高;(黏菌素耐药基因)位于ST10大肠杆菌上的完整前噬菌体上,检测到该前噬菌体与PHAGE__P1最为相似。此外,ST10与ST48型多重耐药大肠杆菌中前噬菌体携带耐药基因概率较高。结果表明在大肠杆菌的前噬菌体上携带耐药基因的概率比较低,且具有特异性。
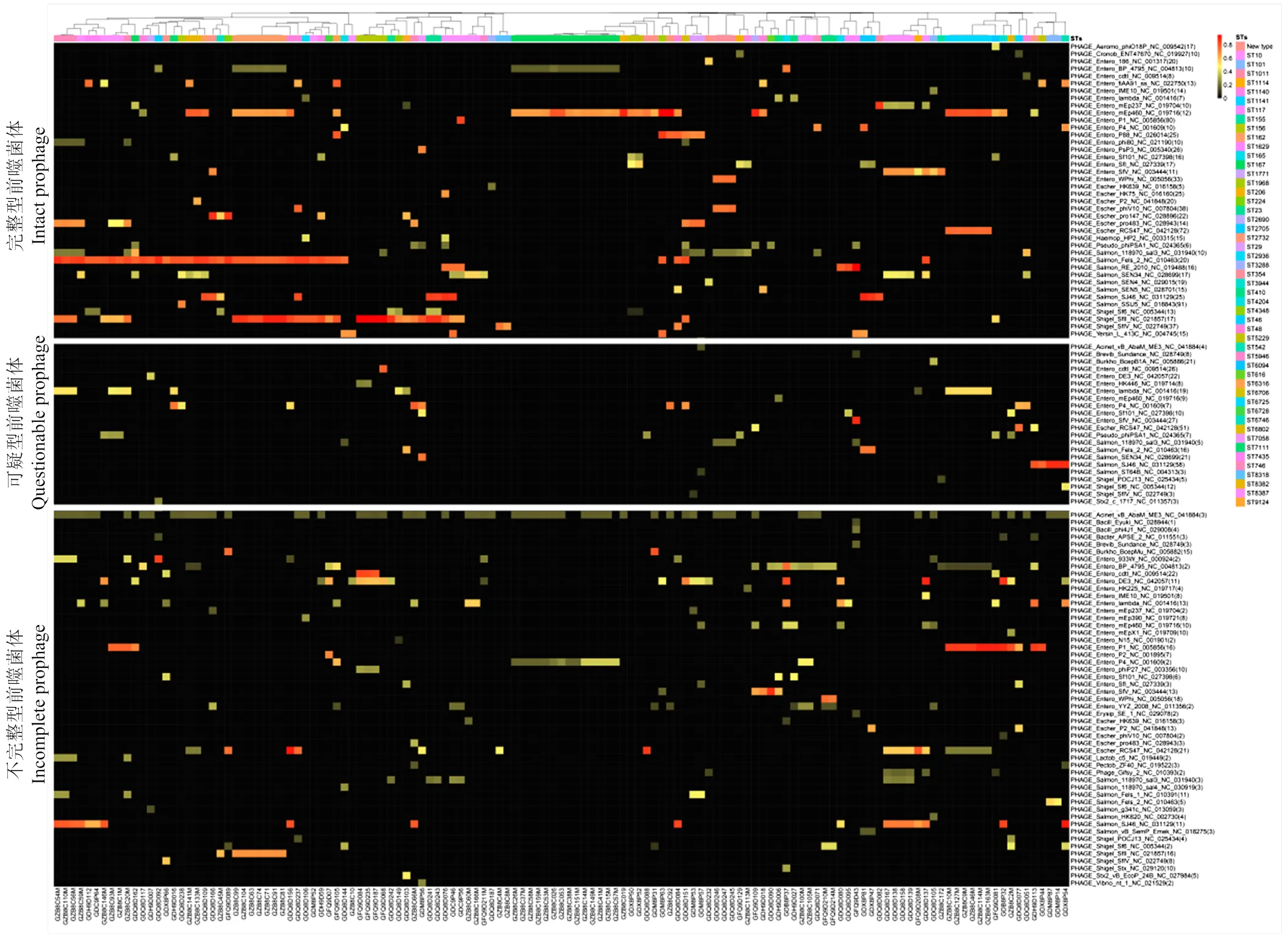
图4 前噬菌体与已知数据库中噬菌体基因组的相似百分比
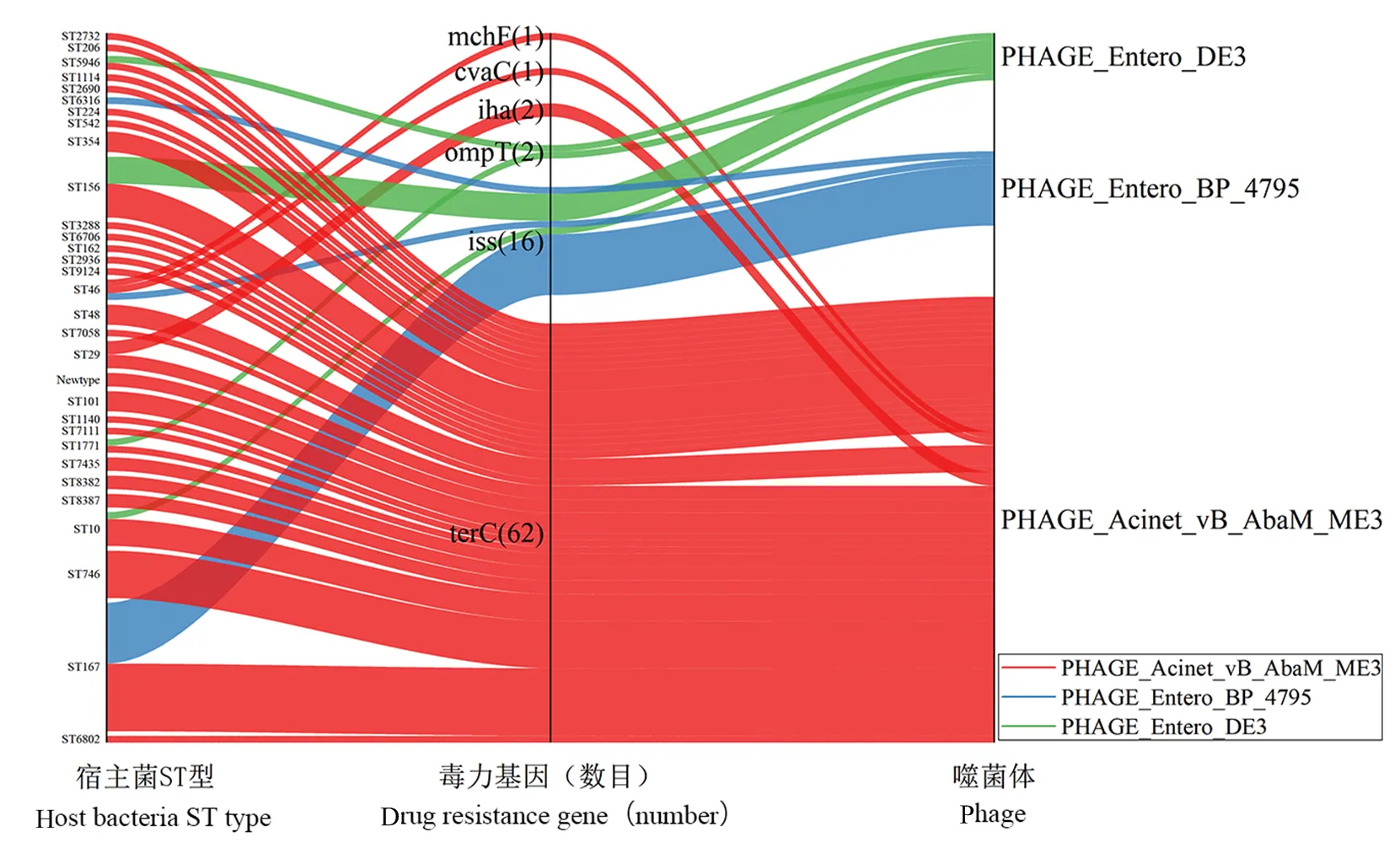
图5 前噬菌体上耐药基因分布图
如图6所示,毒力基因的检测发现在71株前噬菌体携带有6种不同的毒力基因,其中有些菌株携带2种或者3种毒力基因。完整型前噬菌体上分布有17个毒力基因,可疑型前噬菌体上分布6个毒力基因,不完整型前噬菌体上则分布有61个毒力基因。62株前噬菌体携带端粒酶RNA基因,携带的多重耐药大肠杆菌中ST167型占比较大,但总体来说这些前噬菌体对应的多重耐药大肠杆菌ST型分布无明显特征,这些前噬菌体大多为不完整型前噬菌体,这些前噬菌体均与数据库中PHAGE__ vB_AbaM_ME3相似;16株前噬菌体携带血清存活率增加基因带的前噬菌体大多为完整型前噬菌体,这些前噬菌体与PHAGE__BP_ 4795相似度高且基本都分布于ST167型多重耐药大肠杆菌中;外膜蛋白酶、黏附素基因、和ABC转运蛋白基因分别在2、2、1和1株前噬菌体中检出,可通过加工或者降解多种宿主蛋白质使宿主发病,分布于ST1771型和ST5946型多重耐药大肠杆菌中可疑型前噬菌体上与前噬菌体PHAGE__DE3相似;黏附素是生物定植的基础,同时也是细菌致病感染的首要条件,分布于ST29型多重耐药大肠杆菌中不完整型前噬菌体上,与噬菌体PHAGE__vB_AbaM_ME3相似;分布于ST46型多重耐药大肠杆菌中可疑型前噬菌体上,与噬菌体PHAGE__vB_AbaM_ME3相似;分布于ST46型多重耐药大肠杆菌中可疑型前噬菌体上并与噬菌体PHAGE__vB_AbaM_ME3呈现较高相似度。
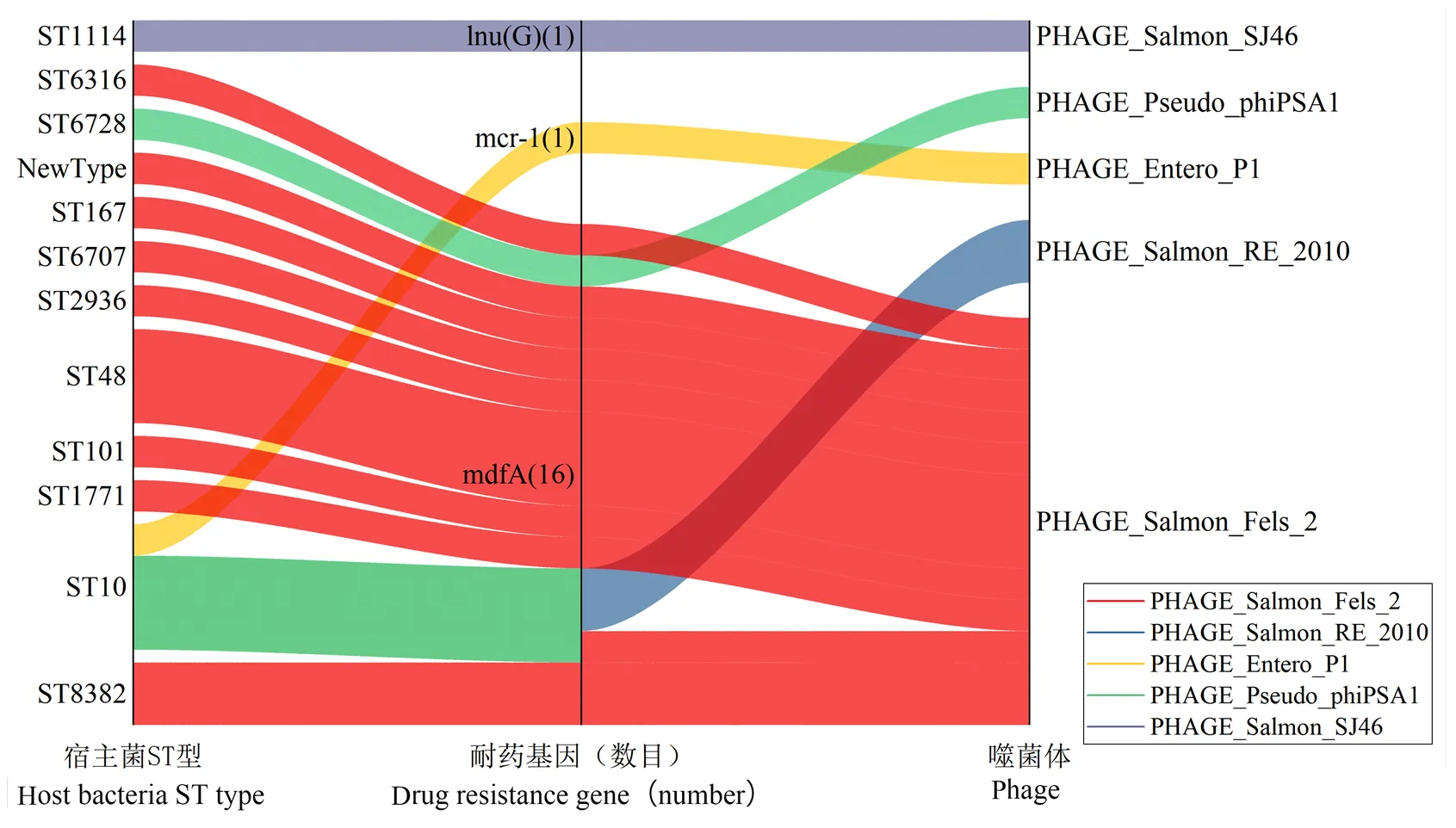
图6 毒力基因在前噬菌体上分布图
2.5 噬菌体的诱导分离
通过丝裂霉素C诱导大肠杆菌,结果显示与对照组比较得到OD600值下降明显(OD600值下降0.3以上认为下降明显)的有110株,占比84.0%;通过双层平板法点板得到噬菌斑的有38株,占比29.0%;分离纯化得到27株,占比20.6%;有11株在纯化过程中未能出现噬菌斑。
3 讨论
目前,已有文献报道噬菌体在细菌间水平基因的转移中发挥着重要作用[35-36],使用宏基因组学分析的方法对环境水流、土壤、人和动物肠道基因组中噬菌体分布的丰度和多样性以及宏噬菌体基因组中ARGs的特性和对其传播的影响进行研究,发现噬菌体对于耐药基因的获得和传播有重要作用[37-39],但是对于细菌基因组中前噬菌体和类前噬菌体的分布、多样性、丰度及其携带耐药基因的特性和对其传播的作用研究非常少。因此本研究对131株大肠杆菌基因组中前噬菌体序列进行了鉴定,针对大肠杆菌中前噬菌体的分布流行特征及其携带ARGs的状况进行分析阐述。
3.1 大肠杆菌中前噬菌体的分布流行特征分析
本研究中,131株大肠杆菌前噬菌体有736个,前噬菌体基因组占其宿主菌的基因组的比例在0.58—5.87之间,在宿主基因组中检测到的前噬菌体与已知噬菌体物种数据库中71种噬菌体以不同的相似百分比匹配上,结果表明前噬菌体在大肠杆菌中分布广泛且具有多样性和高丰度,这与Schmieger和Casjens等[40-41]研究结果相同。该131株大肠杆菌携带大量的ARGs,但检测到的前噬菌体序列上携带的ARGs仅有3种,且概率极小,本研究中的噬菌体基因组上携带耐药基因的数量明显低于相关报道中的环境微生物样品中的宏噬菌体中携带的耐药基因的数量。前噬菌体携带ARGs的概率很低,这可能与细菌的繁殖传代或者与噬菌体共进化过程有关,在不同环境一些特殊的条件下携带耐药基因的细菌与前噬菌体相互作用的概率大大提高,前噬菌体上携带ARGs的能力提高,导致在噬菌体上检出相关的耐药基因,如先前报道的前噬菌体上携带黏菌素耐药基因3.1[42],以及23和NDM-1[43]。然而,目前关于前噬菌体介导耐药基因传播的影响因素目前还不明确,相关的研究有待今后更多的试验来探究。噬菌体编码的毒力因子可以通过细菌分泌到细胞外发挥作用或者在溶原期通过细胞裂解扩散到细胞外起作用[44], 本研究中前噬菌体上携带的毒力基因主要是C和,131株大肠杆菌中有71株大肠杆菌上的前噬菌体携带毒力基因,表明有增强细菌毒力的风险。
3.2 前噬菌体序列比对分析
对于检测到的736个前噬菌体序列经过与NCBI数据库中病毒基因组的比较发现,前噬菌体评分(完整型前噬菌体>可疑型前噬菌体>不完整型前噬菌体)越高,其前噬菌体序列与病毒数据库中基因组序列相似度就越高,但是相似度并没有达到100%完全相似,其原因可能是在细菌和噬菌体共同繁殖进化中,宿主细菌与噬菌体相互适应各自调整的结果[45];也可能是由于数据库中完整的噬菌体基因组序列不足,信息不完整所致。对于温和性噬菌体的诱导和分离,本研究在温和性噬菌体的分离中采用的是大肠杆菌工程菌C600作为宿主菌[46],可以发现OD600下降明显但是分离出单个噬菌斑的概率不高,分析其原因可能是由于诱导过程的不确定性,细菌诱导后点板出斑再次纯化噬菌斑消失原因可能是细菌发生溶原转化,即在噬菌体诱导后细菌再次溶原化[47],可能需要在特定的宿主菌才能出现噬菌斑[48]。
4 结论
131株大肠杆菌中前噬菌体达736个(包括完整和不完整前噬菌体),平均前噬菌体数量为5.6个/株,平均总含量为152.4 kb,占宿主基因组比例大部分在3%。前噬菌体序列中鉴定出3种耐药基因和6种毒力基因,其中包含重要黏菌素耐药基因。前噬菌体在大肠杆菌的基因组中的分布具有多样性且丰度较高,相同ST型下的前噬菌体序列相似度较高。温和性噬菌体的诱导率很高,达到了84.0%。
[1] SALMOND G P C, FINERAN P C. A century of the phage: Past, present and future. Nature Reviews Microbiology, 2015, 13(12): 777-786. doi:10.1038/nrmicro3564.
[2] HOBBS Z, ABEDON S T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiology Letters, 2016, 363(7): 47. doi:10.1093/femsle/fnw047.
[3] MAVRICH T N, CASEY E, OLIVEIRA J, BOTTACINI F, JAMES K, FRANZ C M A P, LUGLI G A, NEVE H, VENTURA M, HATFULL G F, MAHONY J, VAN SINDEREN D. Characterization and induction of prophages in human gut-associatedhosts. Scientific Reports, 2018, 8(1): 12772. doi:10.1038/s41598-018- 31181-3.
[4] TORRES-BARCELÓ C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerging Microbes & Infections, 2018, 7(1): 168. doi:10.1038/s41426-018-0169-z.
[5] SCHMIEGER H, SCHICKLMAIER P. Transduction of multiple drug resistance ofserovar typhimurium DT104. FEMS Microbiology Letters, 1999, 170(1): 251-256. doi:10.1111/j.1574- 6968.1999.tb13381.x.
[6] DAVIES E V, JAMES C E, WILLIAMS D, O'BRIEN S, FOTHERGILL J L, HALDENBY S, PATERSON S, WINSTANLEY C, BROCKHURST M A. Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(29): 8266-8271. doi:10. 1073/pnas.1520056113.
[7] CHEN J, QUILES-PUCHALT N, CHIANG Y N, BACIGALUPE R, FILLOL-SALOM A, CHEE M S J, FITZGERALD J R, PENADÉS J R. Genome hypermobility by lateral transduction. Science, 2018, 362(6411): 207-212. doi:10.1126/science.aat5867.
[8] SONG W, STEENSEN K, THOMAS T. HgtSIM: A simulator for horizontal gene transfer (HGT) in microbial communities. PeerJ, 2017, 5: e4015. doi:10.7717/peerj.4015.
[9] VON WINTERSDORFF C J, PENDERS J, VAN NIEKERK J M, MILLS N D, MAJUMDER S, VAN ALPHEN L B, SAVELKOUL P H, WOLFFS P F. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Frontiers in Microbiology, 2016, 7: 173. doi:10.3389/fmicb.2016.00173.
[10] LERMINIAUX N A, CAMERON A D S. Horizontal transfer of antibiotic resistance genes in clinical environments. Canadian Journal of Microbiology, 2019, 65(1): 34-44. doi:10.1139/cjm-2018-0275.
[11] SHANG Y, LI D, HAO W, SCHWARZ S, SHAN X, LIU B, ZHANG S M, LI X S, DU X D. A prophage and two ICESa2603-family integrative and conjugative elements (ICEs) carrying optrA in. The Journal of Antimicrobial Chemotherapy, 2019, 74: 2876-2879.
[12] HÅFSTRÖM T, JANSSON D S, SEGERMAN B. Complete genome sequence ofintermedia reveals unique genomic features inspecies and phage-mediated horizontal gene transfer. BMC Genomics, 2011, 12(1): 395. doi:10.1186/1471-2164-12-395.
[13] SHAABAN S, COWLEY L A, MCATEER S P, JENKINS C, DALLMAN T J, BONO J L, GALLY D L. Evolution of a zoonotic pathogen: Investigating prophage diversity in enterohaemorrhagicO157 by long-read sequencing. Microbial Genomics, 2016, 2(12): e000096. doi:10.1099/mgen.0.000096.
[14] PLEŠKA M, LANG M, REFARDT D, LEVIN B R, GUET C C. Phage-host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nature Ecology & Evolution, 2018, 2(2): 359-366. doi:10.1038/s41559-017-0424-z.
[15] GOH S, HUSSAIN H, CHANG B J, EMMETT W, RILEY T V, MULLANY P. Phage ϕC2 mediates transduction of Tn6215, encoding erythromycin resistance, betweenstrains. mBio, 2013, 4(6): e00840-e00813. doi:10.1128/mbio.00840-13.
[16] LOHB, CHEN J, MANOHAR P, YU Y, HUA X, LEPTIHN S. A biological inventory of prophages ingenomes reveal distinct distributions in classes, length, and genomic positions. Woqumaid,2020, 11:579802.
[17] COLAVECCHIO A, CADIEUX B, LO A, GOODRIDGE L D. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of thefamily-A review. Frontiers in Microbiology, 2017, 8: 1108. doi:10.3389/fmicb. 2017.01108.
[18] MOHAN RAJ J R, VITTAL R, HUILGOL P, BHAT U, KARUNASAGAR I. T4-likephages from the environment carry blaCTX-M. Letters in Applied Microbiology, 2018, 67(1): 9-14. doi:10.1111/lam.12994.
[19] ZINDER N D, LEDERBERG J. Genetic exchange in. Journal of Bacteriology, 1952, 64(5): 679-699. doi:10.1128/jb.64.5. 679-699.1952.
[20] MAHONY J, VAN SINDEREN D. The impact and applications of phages in the food industry and agriculture. Viruses, 2020, 12(2):210. doi: 10.3390/v12020210.
[21] ANDERSSON D I, HUGHES D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nature Reviews Microbiology, 2010, 8(4): 260-271. doi:10.1038/nrmicro2319.
[22] COLOMER-LLUCH M, IMAMOVIC L, JOFRE J, MUNIESA M. Bacteriophages carrying antibiotic resistance genes in fecal waste from cattle, pigs, and poultry. Antimicrobial Agents and Chemotherapy, 2011, 55(10): 4908-4911. doi:10.1128/aac.00535-11.
[23] COLOMER-LLUCH M, JOFRE J, MUNIESA M. Antibiotic resistance genes in the bacteriophage DNA fraction of environmental samples. PLoS ONE, 2011, 6(3). doi:10.1371/journal.pone.0017549.
[24] LEKUNBERRI I, SUBIRATS J, BORREGO C M, BALCÁZAR J L. Exploring the contribution of bacteriophages to antibiotic resistance. Enviromental Pollution, 2017, 220(pt b): 981-984. doi:10.1016/j. envpol.2016.11.059.
[25] SHOUSHA A, AWAIWANONT N, SOFKA D, SMULDERS F J, PAULSEN P, SZOSTAK M P, HUMPHREY T, HILBERT F. Bacteriophages isolated from chicken meat and the horizontal transfer of antimicrobial resistance genes. Applied and Environmental Microbiology, 2015, 81(14): 4600-4606. doi:10.1128/aem.00872-15.
[26] NOVICK R P, CHRISTIE G E, PENAD S J R. The phage-related chromosomal islands of Gram-positive bacteria. Nature Reviews Microbiology, 2010, 8: 541-551.
[27] VARGA M, KUNTOVÁ L, PANTŮČEK R, MAŠLAŇOVÁ I, RŮŽIČKOVÁ V, DOŠKAŘ J. Efficient transfer of antibiotic resistance plasmids by transduction within methicillin-resistantUSA300 clone. FEMS Microbiology Letters, 2012, 332(2): 146-152. doi:10.1111/j.1574-6968.2012.02589.x.
[28] MAZAHERI NEZHAD FARD R, BARTON M D, HEUZENROEDER M W. Bacteriophage-mediated transduction of antibiotic resistance in enterococci. Letters in Applied Microbiology, 2011, 52(6): 559-564. doi:10.1111/j.1472-765x.2011.03043.x.
[29] DEDRICK R M, JACOBS-SERA D, BUSTAMANTE C A, GARLENA R A, MAVRICH T N, POPE W H, REYES J C, RUSSELL D A, ADAIR T, ALVEY R, et al. Prophage-mediated defence against viral attack and viral counter-defence. Nature Microbiology, 2017, 2: 16251.
[30] TRAN P M, FEISS M. φSa3mw Prophage as a Molecular Regulatory Switch ofβ-Toxin Production. 2019, 201(14): e00766-18. doi: 10.1128/JB.00766-18.
[31] OGATA S, SUENAGA H, HAYASHIDA S. A temperate phage of. Applied and Environmental Microbiology, 1985, 49(1): 201-204. doi:10.1128/aem.49.1.201-204.1985.
[32] JOFRE J, MUNIESA M. Bacteriophage isolation and characterization: phages of. Methods in Molecular Biology (Clifton, N J), 2020, 2075: 61-79. doi:10.1007/978-1-4939-9877-7_4.
[33] ARNDT D, MARCU A, LIANG Y, WISHART D S. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Briefings in Bioinformatics, 2019, 20(4): 1560-1567. doi:10.1093/bib/bbx121.
[34] WANG D, LIANG H, CHEN J, MOU Y, QI Y. Structural and environmental features of novel mdfA variant and mdfA genes in recombinant regions of. Microbial Drug Resistance (Larchmont, N Y), 2014, 20(5): 392-398. doi:10.1089/mdr.2013.0201.
[35] BATTAGLIOLI E J, BAISA G A, WEEKS A E, SCHROLL R A, HRYCKOWIAN A J, WELCH R A. Isolation of generalized transducing bacteriophages for uropathogenic strains of. Applied and Environmental Microbiology, 2011, 77(18): 6630- 6635. doi:10.1128/aem.05307-11.
[36] ZHANG A, CALL D R, BESSER T E, LIU J, JONES L, WANG H, DAVIS M A. Β-lactam resistance genes in bacteriophage and bacterial DNA from wastewater, river water, and irrigation water in Washington State. Water Research, 2019, 161: 335-340. doi:10.1016/j.watres.2019. 06.026.
[37] CALERO-CÁCERES W, YE M, BALCÁZAR J L. Bacteriophages as environmental reservoirs of antibiotic resistance. Trends in Microbiology, 2019, 27(7): 570-577. doi:10.1016/j.tim.2019.02.008.
[38] GARIN-FERNANDEZ A, PEREIRA-FLORES E, GLÖCKNER F O, WICHELS A. The North Seaviral: Occurrence and distribution of North Sea bacteriophages. Marine Genomics, 2018, 41: 31-41. doi:10.1016/j.margen.2018.05.004.
[39] WENDLING C C, REFARDT D, HALL A R. Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic- resistance genes peak in different environments. BioRxiv, 2020. DOI:10.1101/2020.03.13.990044. doi:10.1101/2020.03.13.990044.
[40] LEKUNBERRI I, VILLAGRASA M, BALCÁZAR J L, BORREGO C M. Contribution of bacteriophage and plasmid DNA to the mobilization of antibiotic resistance genes in a river receiving treated wastewater discharges. The Science of the Total Environment, 2017, 601/602: 206-209. doi:10.1016/j.scitotenv.2017.05.174.
[41] WANG M, XIONG W, LIU P, XIE X, ZENG J, SUN Y, ZENG Z. Metagenomic insights into the contribution of phages to antibiotic resistance in water samples related to swine feedlot wastewater treatment. Frontiers in Microbiology, 2018, 9: 2474. doi:10.3389/ fmicb.2018.02474.
[42] PAN Y, FANG Y, FENG Y, LYU N, CHEN L, LI J, XU X, ZHU B, HU Y. Discovery of mcr-3.1 gene carried by a prophage located in a conjugative IncA/C2 plasmid from aCholeraesuis clinical isolate. The Journal of Infection, 2021, 82(3): 414-451. doi:10.1016/ j.jinf.2020.09.036.
[43] LOH B, CHEN J, MANOHAR P, YU Y, HUA X, LEPTIHN S. A biological inventory of prophages ingenomes reveal distinct distributions in classes, length, and genomic positions. Frontiers in Microbiology, 2020, 11: 579802.
[44] HSU B B, WAY J C, SILVER P A. Stable neutralization of a virulence factor in bacteria using temperate phage in the mammalian gut. mSystems, 2020, 5.
[45] MOLINA F, SIMANCAS A, TABLA R, GÓMEZ A, ROA I, REBOLLO J E. Diversity and local coadaptation ofand coliphages from small ruminants. Frontiers in Microbiology, 2020, 11: 564522. doi:10.3389/fmicb.2020.564522.
[46] FRY B A. Conditions for the infection ofwith lambda phage and for the establishment of lysogeny. Journal of General Microbiology, 1959, 21: 676-684.
[47] IMAMOVIC L, BALLESTÉ E, MARTÍNEZ-CASTILLO A, GARCÍA- ALJARO C, MUNIESA M. Heterogeneity in phage induction enables the survival of the lysogenic population. Environmental Microbiology, 2016, 18(3): 957-969. doi:10.1111/1462-2920.13151.
[48] RUIZ-CRUZ S, PARLINDUNGAN E, ERAZO GARZON A, ALQARNI M, LUGLI G A. Lysogenization of a lactococcal host with three distinct temperate phages provides homologous and heterologous phage resistance. Microorganisms, 2020, 8(11). doi.org/10.3390/ microorganisms8111685.
Distribution Characteristics of Prophage in Multidrug Resistantas well as Its Induction and Isolation
LIU Jiao, LIU Chang, CHEN Jin, WANG MianZhi, XIONG WenGuang, ZENG ZhenLing
South China Agriculture University/Guangdong Provincial Key Laboratory of Veterinary Pharmaceutics Development and Safety Evaluation/National Risk Assessment Laboratory for Antimicrobial Resistance of Animal Original Bacteria, Guangzhou 510642
【】This study investigated the distribution characteristics of prophage in multi-drug resistant, induction and isolation, as well as the prevalence of drug resistance and virulence genes in prophage, so as to provide a scientific basis for the study of prophage-mediated resistance genes in the spread of bacteria. 【】 131 multi-drug resistantisolating from poultry origin in Guangdong Province from 2018 to 2019 were selected in the laboratory for nucleic acid extraction and whole-genome sequencing. The results of second-generation sequencing were assembled and spliced into a whole-genome sequence and uploaded to the phage. The PHASTER network database was compared and analyzed with the existing phage genome sequences in the database. Drug resistance genes and virulence genes were compared on the CGE database, and then the distribution of drug resistance genes and virulence genes on the prophage were obtained. The mild phage was induced by mitomycin C, separated and purified by using the double-layer plate method. 【】 The results of the drug sensitivity test of 131 strains ofshowed that the drug resistance rates of ampicillin, tetracycline, florfenicol and compound trimethoprim were all more than 90%, followed by cephalosporin antibiotics, gentamicin, ciprofloxacin, meropenem and colistin with all around 50%, and the resistance rate of tigecycline reached 0.2%. All strains showed multi-drug resistance, and they were all multi-drug resistant. A total of 736 prophage fragments were detected in 131 strains of multi-drug resistant, including 329 complete prophage, 66 suspicious phages and 341 incomplete phage, which matched with 40, 20 and 52 known database phage species in different percentages, respectively. The gene sequence of the complete prophage showed that it matched the known phage species better, and the sequence similarity was the highest, with an average of 58.53%. The average number of prophages in 131 strains ofwas 5.6, and the average total content was 152.4 kb. Prophage genome accounted for 0.58% to 5.87% of its host genome, with 3.0% being the dominant. The length of the prophage genome ranged from 2.8 to 107.9 kb, and the 13.0 kb prophage had the highest frequency, accounting for 9.1% of all prophages. CGE comparison results showed that the genomes of 131 strains of multi-drug-resistantdetected resistance genes(A),(G) and(A),(G) and mcr-1 were 16, 1, and 1, respectively. 71 strains of multi-drug resistantprophage carried 6 different virulence genes, and some strains carried 2 or 3 virulence genes. There were 62 prophages carrying the telomerase RNA gene, 16 prophages carrying the serum survival increasing gene, and the outer membrane protease, among which the adhesin gene, thegene and the ABC transporter genewere at 2, 2, 1, and 1, respectively. Mcr-a gene were detected in prophage of 1 strain multi-drug resistant. The(A) gene andgene were the most common resistance genes and virulence genes in prophage, respectively. The results of mild phage induction experiments showed that the success rate of prophage induction was 84.0%, but the probability of plaque appearance was still relatively low. 【】Prophages were widely distributed in multi-drug resistantand carried a variety of resistance genes and virulence genes. Mild phages had a high induction rate, and have the risk of horizontal transmission of resistance genes and virulence genes, and need to be strengthened and sustained monitor.
prophage;; induction; distribution characteristics; resistance genes

10.3864/j.issn.0578-1752.2022.07.017
2021-02-06;
2021-09-30
国家自然科学基金面上项目(31872524)、广东省普通高校省级重大科研项目(2017KZDXM006)
刘教,E-mail:1766612575@qq.com。通信作者曾振灵,E-mail:zlzeng@scau.edu.cn
(责任编辑 林鉴非)
