沙棘枝枯病的定量检测体系构建和应用
2022-04-12孙思雨胡建忠郝凯强吴元华
陈 悦,孙思雨,胡建忠,郝凯强,于 曼,吴元华,夏 博
(1.沈阳农业大学,辽宁 沈阳 110866;2.水利部沙棘开发管理中心,北京 100038)
沙棘Hippophae rhamnoides是一种多年生落叶灌木,广泛分布于欧亚大陆,我国从东北、华北到西北、西南均是沙棘主栽地区[1]。我国现有沙棘资源200 余万hm2,沙棘加工企业200 余家,年产值约70 亿元[2]。沙棘适生性强,可以通过硬枝扦插、嫩枝扦插和嫩芽扦插等途径进行快速繁育[3],适宜在我国西部荒漠化地区大面积栽培,对水土保持、防风固沙具有重要作用,兼具重要的经济价值和生态价值[4]。
沙棘枝枯病又被称为干缩病,是世界范围内严重危害沙棘的毁灭性病害之一。20世纪60年代该病在俄罗斯大面积爆发,目前已在世界沙棘主栽区广泛流行[5]。在我国辽宁、甘肃及陕西等沙棘主要种植区均有沙棘枝枯病危害的报道[6]。最近有研究结果表明,引起黑龙江省沙棘枝枯病的病原菌为拟枝孢镰刀菌Fusarium sporotrichioides[7]。拟枝孢镰刀菌是一类具有广泛寄主的病原真菌,可以侵染马铃薯、紫花苜蓿、大豆等多种经济作物[8-10],能够在植物病残体和土壤中越冬越夏,引起玉米、薰衣草等多种植物枯萎病、腐烂病的发生[11-13]。发病植株上部叶片提前褪绿并大量脱落,在枝上形成凹陷病斑,随后病斑连结成片,整个植株的枝条干枯皱缩,严重感病的植株出现沙棘果实提早脱落的现象,该病最终导致沙棘林木大面积死亡[14]。
实时荧光定量PCR 技术(quantitative realtime PCR)是基于DNA/RNA 探针技术的植物病原菌鉴定方法,在PCR 反应体系中加入荧光基团,利用荧光信号的累积来实时监测整个PCR 反应过程,最后再通过已建立的标准曲线对未知模板进行定量分析,与传统诊断方法相比具有高度特异性[15],被广泛应用于基因表达的定量分析[16-17]。传统PCR 方法的灵敏度低,且采用传统PCR 方法无法实现对环境和土壤样品的高效定量检测[18],而实时荧光定量PCR 具有较高的检测灵敏性,兼顾定性检测和定量检测,采用该技术能够较为准确地确定初始拷贝数[19-20],该技术已经被广泛应用于定量检测土壤和枝条病残体中微生物含量[21]。实时荧光定量PCR 技术在拟枝孢镰刀菌检测方面的应用研究鲜见报道。本研究中针对拟枝孢镰刀菌建立实时荧光定量PCR 检测体系,旨在从基因水平为快速定量检测拟枝孢镰刀菌提供一种有效方法,对沙棘枝枯病的早期诊断、定量检测和病害防控提供参考。
1 材料与方法
1.1 供试材料
1.1.1 供试菌株
供试菌株为单孢纯化后的G1-5-1 拟枝孢镰刀菌F.sporotrichioides、 核盘菌Sclerotinia sclerotiorum、 炭疽菌Colletotrichumsp.、 根肿菌Plasmodiophora brassicae、丝核菌Rhizoctoniasp.、尖孢镰刀菌Fusarium oxysporum、层出镰刀菌Fusarium proliferatum、木贼镰刀菌Fusarium equisetum、 三线镰刀菌Fusarium tricinctum、赤霉菌Gibberellasp.、 拟茎点霉Phomopsis cauliflorum,现存于沈阳农业大学植物保护学院病毒室。
1.1.2 供试枝条样本和土壤样本
2020年5—7月,在甘肃省庆阳市采集17 份枝条样本、4 份沙棘根际土壤样本,共21 份样本,其详细信息见表1。
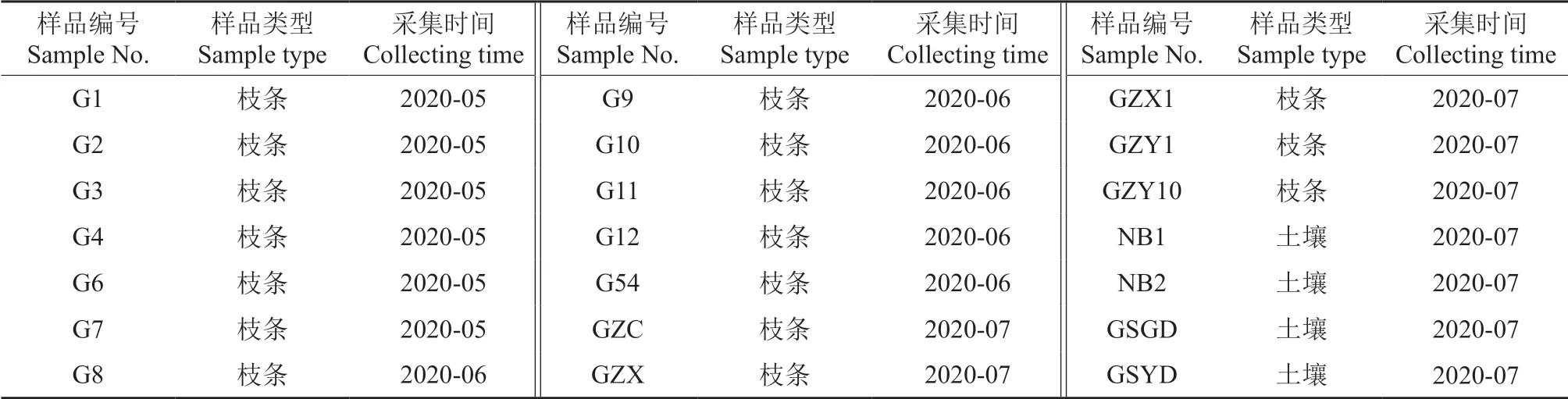
表1 供试沙棘枝条和土壤样品Table 1 Test branch and soil samples
1.2 试剂与仪器
试剂:NaCl、无水乙醇和蔗糖购自国药公司;胰蛋白胨、酵母提取物、琼脂糖和卡那霉素购自索莱宝公司;胶回收试剂盒(全式金);DNA secure新型植物基因组DNA 提取试剂盒(Tiangen);土壤微生物基因组DNA 提取试剂盒(Qiagen);真菌基因组快速抽提试剂盒(生工,上海);其他未标注试剂均购自Vazyme 公司。
仪器:紫外凝胶成像仪(Bio-Rad)、PCR 扩增仪StepOnePlus ™(Thermofisher)、Nanodrop紫外分光光度计(Thermofish)。
1.3 试验方法
1.3.1 基因组DNA 制备
1)病原菌基因组DNA 的提取。在单孢纯化后的菌落上打取直径为5 mm 的菌饼,将菌饼置于PD 培养液中,封好封口膜,在摇床(25 ℃,110 r/min)上摇培5 d 后,用已灭菌的滤纸过滤培养液,将过滤出的菌丝用无菌水冲洗3 次放于50 mL 离心管中,在冻干机中冻干3 h。参照真菌基因组快速抽提试剂盒说明书提取病原菌基因组DNA。取20 mg 干燥的子实体或菌丝放入研钵中,倒入少量液氮,迅速研磨成粉末,加入到1.5 mL离心管中。加入400 µL Buffer Digestion 和4 µL β-巯基乙醇,振荡混匀。65 ℃水浴1 h,至细胞完全裂解。加入200 µL Buffer PF,充分颠倒混匀,在冰箱(-20 ℃)中放置5 min。室温条件下,10 000 r/min 离心5 min,将上清液(500 ~550 µL)转移到新的1.5 mL 离心管中。加入等体积的异丙醇,颠倒5 ~8 次使之充分混匀,室温条件下放置2 ~3 min。室温条件下,10 000 r/min 离心5 min,弃上清液。加入1 mL 75%的乙醇,颠倒漂洗1 ~3 min,10 000 r/min 离心2 min,弃上清,重复1 次该操作步骤。开盖,室温条件下倒置5 ~10 min,至残留的乙醇完全挥发。得到的DNA 用50~100 µL TE Buffer溶解,并在-20 ℃条件下保存。
2)植物基因组DNA 的提取。参照DNA secure 新型植物基因组DNA 提取试剂盒说明书提取植物基因组DNA。取植物干组织20 mg 放入研钵中,倒入少量液氮,充分碾磨,加入400 µL 缓冲液LP1 和6 µL RNase A(10 g/L),放入离心管中,旋涡振荡1 min,室温条件下放置10 min。加入130 µL 缓冲液LP2,充分混匀,旋涡振荡1 min。12 000 r/min 离心5 min,将上清液移至新的离心管中。加入1.5 倍体积的缓冲液LP3,立即充分振荡15 s,使之混匀,可能会出现絮状沉淀。将所得溶液和絮状沉淀均加入吸附柱CB3 中,1 200 r/min 离心30 s,弃废液,将吸附柱CB3 放入收集管中。向吸附柱CB3 中加入600 µL 漂洗液PW,1 200 r/min 离心30 s,弃废液,将吸附柱CB3 放入收集管中,重复该操作步骤多次。然后1 200 r/min 离心2 min,弃废液。吸附柱CB3 在室温条件下放置2 min,彻底晾干残余的漂洗液。将吸附柱CB3 转入新的1.5 mL 离心管中,向吸附膜中间位置悬空滴加50 ~200 µL 洗脱缓冲液TE,在室温条件下放置2 min,12 000 r/min 离心2 min,将溶液收集到离心管中。
3)土壤微生物基因组DNA 的提取。参照土壤微生物基因组DNA 提取试剂盒(Qiagen)说明书提取土壤微生物基因组DNA。向Power Bead Tube 中加入0.25 g 土壤样品,轻轻搅拌。加入60 μL 的C1,涡旋振荡数秒。24 位旋涡适配器水平固定,高速涡旋10 min,12 000 r/min 离心30 s。将上清液移到2 mL 收集管中,加入250 μL C2 涡旋5 s,在2 ~8 ℃条件下静置5 min。12 000 r/min离心1 min,将600 μL 上清液移到2 mL 收集管中。加入200 μL C3 并涡旋5 min,在2 ~8 ℃条件下静置5 min。12 000 r/min 离心30 s,将750 μL 上清液移到2 mL 收集管中。摇匀C4,加入1 200 μL上清液,涡旋5 s。取该溶液675 μL 加入MB 旋转柱离心管中,12 000 r/min 离心30 s,弃掉流出液,重复该操作步骤多次,处理所有样品。添加500 μL 的C5,12 000 r/min 离心30 s。弃掉流出液,12 000 r/min 再次离心1 min。将MB 旋转柱放入2 mL收集管中,加入100 μL的C6到白色滤膜中央。在室温条件下12 000 r/min 离心30 s,弃掉MB 旋转柱,试管中的DNA 可用于下一步试验。
1.3.2 常规PCR 反应
根据荧光定量PCR 引物的设计原则,使用Primer 5 软件进行引物设计。所设计引物序列为QS-2F(5′-CATCATTCGAATCGCTCTCA),和QS-2R(5′-GGGTATGAGCCCCACTATCA),由上海生工生物公司(长春)合成。
以总DNA 为模板,加入所设计实时荧光定量PCR 特异性引物,对目标片段进行扩增。反应体系为10 µL 2×Taq Master Mix、8.2 µL ddH2O、1 µL 总DNA、0.4 µL primer-F(10 μmol/L)、0.4 µL primer-R(10 μmol/L),反应体系总体积为20 µL。PCR 反应条件:94 ℃预变性3 min;94 ℃变性15 s,60 ℃退火15 s,72 ℃延伸30 s,30 ~35个循环;72 ℃延伸5 min,4 ℃保存。
1.3.3 目的片段载体连接与热激转化
1)胶回收与载体连接。参照天根公司提供的胶回收试剂盒说明书进行切胶回收。向吸附柱CA2 中加入500 µL 平衡液BL,12 000 r/min 离心1 min,倒掉收集管中的废液,将吸附柱重新放回收集管中。将单一的目的DNA 条带从琼脂糖凝胶中切下,放入干净的离心管中,称取其质量。向胶块中加入等体积的PN 溶液,50 ℃水浴放置,其间不断温和地上下翻转离心管,以确保胶块充分溶解。将所得溶液加入吸附柱CA2 中,在室温条件下放置2 min,12 000 r/min 离心30 ~60 s,倒掉收集管中的废液,将吸附柱CA2 放入收集管中。向吸附柱CA2 中加入600 µL 漂洗液PW,12 000 r/min 离心30 ~60 s,弃废液,将吸附柱CA2 放入收集管中,重复1 次该操作步骤。将吸附柱CA2 放回收集管中,12 000 r/min 离心2 min,除尽漂洗液后,在室温条件下放置晾干;再将吸附柱CA2 放到离心管中,向吸附膜中间位置悬空滴加适量洗脱缓冲液EB,在室温条件下放置2 min。12 000 r/min 离心2 min,收集DNA溶液。连接体系为4 µL 回收产物DNA、0.5 µL pEASY-T1 simple 载体、无菌水0.5 µL,总体积5 µL,反应5 min(25 ℃)。
2)热激转化。将上述产物加入50 µL trans1-T1 感受态细胞中,冰浴20 ~30 min。42 ℃金属浴热激30 s,反应结束后立即将产物冰浴2 min。将250 µL 液体LB 培养基加入上述体系中,在37 ℃条件下200 r/min 培养1 h。3 000 r/min 离心1 min,弃掉150 µL,轻弹混匀,取50 µL 均匀涂布到含50 mg/L 卡那霉素的LB 固体培养基上。
3)阳性菌落检测和测序。挑选单菌落加入到1 mL 含50 mg/L 卡那霉素的液体LB 培养基中,在37 ℃条件下200 r/min 培养7 ~8 h。取1 µL 菌液作为模板加入到PCR(20 µL 体系)预混液中,进行PCR 反应。将检测到的阳性克隆质粒送到上海生工生物公司(长春)测序。
4)质粒提取。按照天根公司提供的试验方法进行质粒提取。向吸附柱CP3 中加入500 µL 平衡液BL,12 000 r/min 离心1 min,弃废液,将吸附柱重新放回收集管中。取1 ~5 mL 过夜培养的菌液,加入离心管中,12 000 r/min 离心1 min,吸除上清液。向留有菌体沉淀的离心管中加入250 µL的P1 溶液,使用涡旋振荡器使悬浮细菌彻底沉淀。向离心管中加入250 µL 的P2 溶液,温和地上下翻转6 ~8 次,使菌体充分裂解。再加入350 µL 的P3 溶液,温和地上下翻转6 ~8 次,使之充分混匀,出现白色絮状沉淀。12 000 r/min 离心10 min。将收集的上清液转移到吸附柱CP3 中,尽量不要吸出沉淀。12 000 r/min 离心30 ~60 s,弃废液,将吸附柱CP3 放入收集管中。向吸附柱CP3 中加入500 µL去蛋白液PD,12 000 r/min离心30~60 s,弃废液,将吸附柱CP3 重新放回收集管中。再加入600 µL 漂洗液PW,12 000 r/min 离心30 ~60 s,弃废液,将吸附柱CP3 放入收集管中,重复该操作步骤多次。将吸附柱CP3 放入收集管中,12 000 r/min 离心2 min。将吸附柱CP3 置于离心管中,向吸附膜的中间部位滴加50 ~100 µL 洗脱缓冲液EB,在室温条件下放置2 min,12 000 r/min离心2 min,将质粒溶液收集到离心管中。
1.3.4 实时荧光定量PCR 检测
实时荧光定量PCR 反应体系为10 µL 2×ChamQ Universal SYBR qPCR Master Mix、0.4 µL Primer 1(10 μmol/L)、0.4 µL Primer 2(10 μmol/L)、2 µL Template DNA、7.2 µL ddH2O,反应体系总体积为20 µL。反应条件:95 ℃预变性30 s;95 ℃变性10 s,56 ℃退火30 s,72 ℃延伸1 min,40个循环。
1)引物特异性检测。选取拟枝孢镰刀菌、层出镰刀菌和三线镰刀菌等自沙棘枝条和根际土壤分离的真菌DNA 作为模板,用ddH2O 作为阴性对照,进行实时荧光定量PCR,检测引物的特异性。
2)实时荧光定量PCR 灵敏性检测。将提取的拟枝孢镰刀菌的DNA 进行梯度稀释,使每微升模板的拷贝数为103~108。以梯度稀释质粒DNA作为模板,以ddH2O 作为阴性对照,进行实时荧光定量PCR 反应,检测实时荧光定量PCR 的灵敏性。由仪器内置程序绘制标准曲线,标准曲线的横坐标为样品拷贝数的对数,纵坐标为反应循环数。
N=(C×6.02×1023)/[(L+L′)×660]。
式中:N为每微升模板的质粒拷贝数;C为质粒体积浓度;L为载体长度;L′为片段长度。
3)枝条和土壤样品检测。将所提取的枝条和土壤微生物基因组DNA 样品进行实时荧光定量PCR 扩增反应。
2 结果与分析
2.1 拟枝孢镰刀菌EF-1α 质粒的构建
将构建好的拟枝孢镰刀菌EF-1α 质粒,以实时荧光定量PCR 特异性引物(QS-2F、QS-2R)进行菌落PCR,使用1.4%琼脂糖进行凝胶电泳检测,结果如图1所示。由图1 可见,阳性菌落条带明显,泳道4(CK)无条带。将测序结果进行序列比对,发现同源性高达100%,说明EF-1a质粒构建成功。
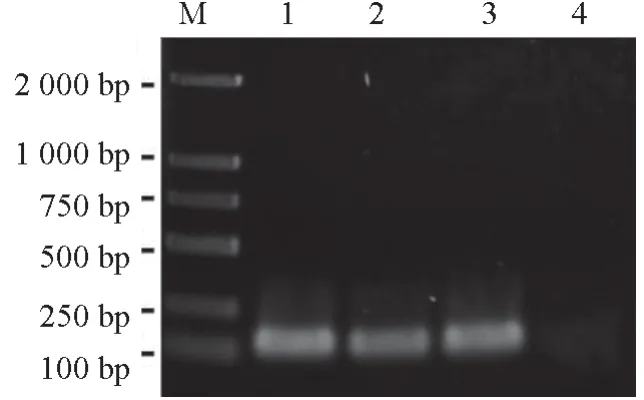
图1 拟枝孢镰刀菌EF-1α 质粒特异性检测结果Fig.1 Specific detection result of F.sporotrichioides EF-1α fragment plasmid
2.2 实时荧光定量PCR 引物的特异性
PCR 引物特异性检测结果如图2所示。由图2 可见,所设计的实时荧光定量PCR 特异性引物(QS-2F、QS-2R)以拟枝孢镰刀菌为DNA 模板能进行有效扩增,且PCR 呈阳性反应,其他供试菌株的DNA 模板及阴性对照均未检测到条带,证明该引物对拟枝孢镰刀菌具有较高的特异性。

图2 实时荧光定量PCR 引物特异性的电泳检测结果Fig.2 PCR primer specific detection
2.3 拟枝孢镰刀菌实时荧光定量PCR 的灵敏性
提取拟枝孢镰刀菌EF-1α 质粒DNA 后,分别稀释成不同质粒DNA 模板浓度,使每微升模板的拷贝数为103~108,进行拟枝孢镰刀菌实时荧光定量PCR 扩增(图3)和常规PCR 扩增。
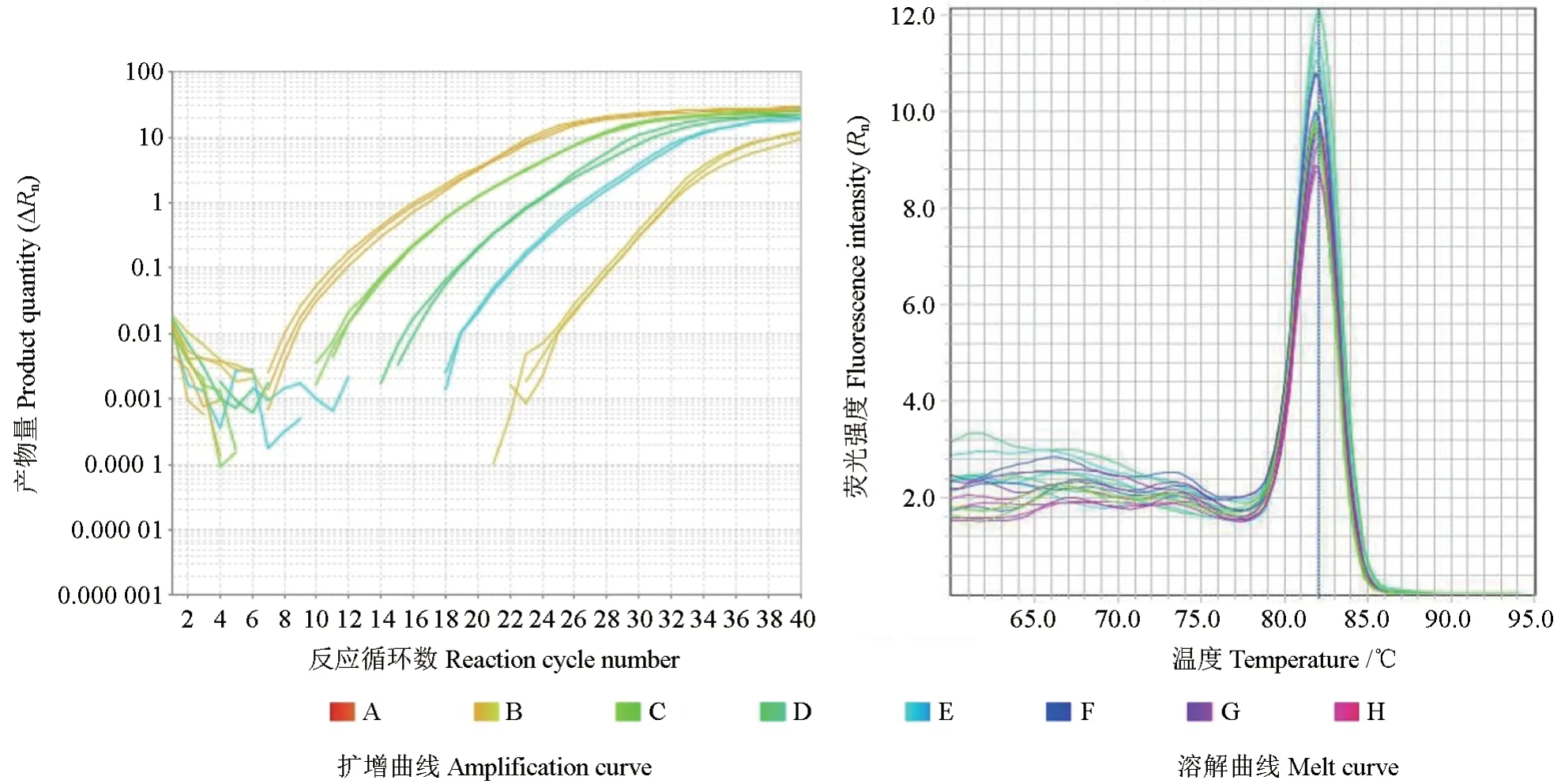
图3 拟枝孢镰刀菌EF-1α 质粒实时荧光定量PCR 扩增结果Fig.3 Amplification result of quantitative real-time PCR for F.sporotrichioides EF-1α fragment plasmid
拟枝孢镰刀菌实时荧光定量PCR 标准曲线如图4所示。由图4 可见,反应循环数和DNA 浓度的对数呈现线性关系。
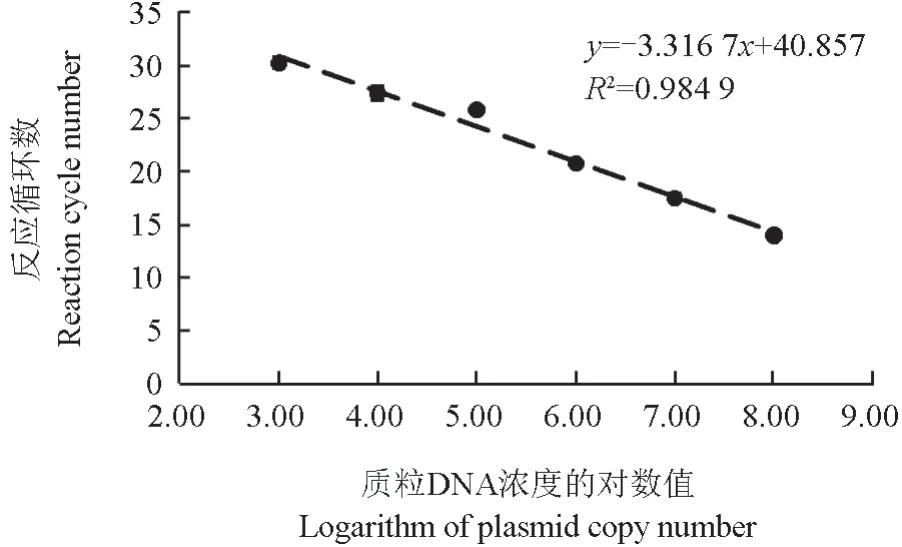
图4 拟枝孢镰刀菌实时荧光定量PCR 标准曲线Fig.4 Standard curve of quantitative real-time PCR for F.sporotrichioides
应用常规PCR 扩增结果进行电泳,结果如图5所示。由图5 可见,以质粒作为模板且每微升的拷贝数为108~104时,有明显条带;在每微升的拷贝数为103时,能观察到微弱条带;在每微升的拷贝数为102、10 时及阴性对照中均未检测到条带。进行实时荧光定量PCR 扩增时,能够有效检测每微升的拷贝数为103。表明所建立的实时荧光定量PCR 方法的灵敏性优于常规PCR。

图5 普通PCR 检测不同浓度拟枝孢镰刀菌EF-1α 质粒DNA 的电泳结果Fig.5 The electrophoresis diagram of ordinary PCR for different concentrations of plasmid DNA of F.sporotrichioides
2.4 枝条和土壤DNA 样品的拟枝孢镰刀菌实时荧光定量PCR 定量检测
将枝条和土壤DNA 样品进行拟枝孢镰刀菌实时荧光定量PCR 扩增,结果表明在25 份枝条样品中有6 份存在拟枝孢镰刀菌,在12 份土壤样品中有1 份存在拟枝孢镰刀菌。将检测结果代入拟枝孢镰刀菌实时荧光定量PCR 标准曲线,结果如图6所示。由图6 可见:每微升所提取枝条DNA样品的拟枝孢镰刀菌拷贝数为103.75~104.15,即每克枝条样品的拟枝孢镰刀菌拷贝数为6×104.75~6×105.15;每微升所提取GSGD 土壤DNA 样品的拟枝孢镰刀菌拷贝数是103.82,即每克土壤样品的拟枝孢镰刀菌拷贝数为6×104.82。
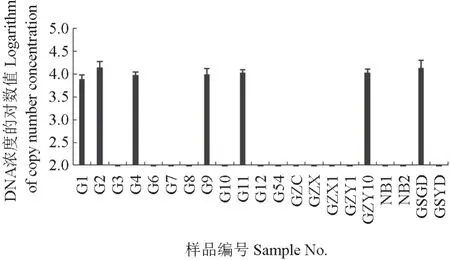
图6 枝条和土壤样品中拟枝孢镰刀菌数量的实时荧光定量PCR 定量检测结果Fig.6 Quantitative real-time PCR quantitative detection of the expression of F.sporotrichioides in branches and soil
3 结论与讨论
沙棘枝枯病是由拟枝孢镰刀菌引起的系统性病害,有必要建立一套有效的拟枝孢镰刀菌检测体系进行沙棘枝枯病的早期防控。本研究中通过设计特异性引物,优化了一种精准检测沙棘枝枯病的方法—拟枝孢镰刀菌实时荧光定量PCR 法。本研究中所用引物是基于翻译延伸因子(EF-1α)设计的[22],可以特异性扩增拟枝孢镰刀菌,能够较好地区分拟枝孢镰刀菌、尖孢镰刀菌、三线镰刀菌以及其他多种致病菌,同时能够定性定量检测沙棘枝干和土壤的病原菌密度。本研究中建立了拟枝孢镰刀菌质粒检测标准曲线,使用该曲线得到的每微升DNA 样本的检测拷贝数是103~108。通过对21 份枝条和土壤样品进行检测,在7份样品中发现荧光信号,进一步证明了该方法的实用性和可操作性。
本研究中采用的拟枝孢镰刀菌实时荧光定量PCR 检测方法可替代传统的稀释平板计数法,具有易操作、特异性强和灵敏度高等优点,该方法有利于研究病原菌在沙棘植物组织和土壤中的分布。
目前,常规PCR 已被应用于生态监测、植物病害诊断和病原物鉴定等方面[18],但是该方法仅适用于植物枝干和土壤内含病原菌的鉴定,不能进行定量检测[8]。土壤中病原菌的传统检测方法操作复杂、灵敏度低且检测时间长[21]。实时荧光定量PCR 技术既有常规PCR 扩增的高灵敏性,且兼备DNA 杂交的高特异性和光谱技术的高精确定量,既弥补了传统PCR 检测的缺点,又不会对环境造成污染[23]。该技术的采用大大降低了对植物枝干、空气及土壤中病原菌的定性和定量检测难度。例如,该技术在小麦叶片中条锈病菌和白粉病菌的检测[24-25]及水稻种子中恶苗病菌的定量检测[26]中均得到了较好应用[27]。本研究中仅针对甘肃地区沙棘枝枯病样品的病原菌拟枝孢镰刀菌进行了定量检测,还应使用其他地区的样品对该检测方法进行验证和优化。
