废弃塑料化学回收及升级再造研究进展
2022-04-12陈欢万坤牛波张亚运龙东辉
陈欢,万坤,牛波,张亚运,龙东辉
(华东理工大学化工学院,上海 200237)
塑料是以小分子单体为原料,通过加聚或缩聚反应聚合而成的高分子化合物。自20世纪50 年代塑料制品推广以来,由于其来源广泛、使用便捷、性能优良、易于加工、价格低廉等特点,被广泛应用于包装、建筑、汽车制造、医疗器械等诸多国民经济重要领域。
迄今为止人类已经生产了超过80亿吨的塑料,其中大约80%被丢弃。2019 年全球塑料产量已达3.68亿吨,预计2050年将超过5亿吨,并且仍在以3%~4%的年增长速度增加。目前,我国仍是世界上最大的塑料生产国和消费国,2020 年我国塑料制品产量约为7603 万吨,表观消费量约为1.3 亿吨。预计到2025 年,我国产生的塑料垃圾将超过1700万吨,成为世界上最大的塑料垃圾生产国。
由于塑料的使用耐久性和耐分解性,废弃塑料在自然界完全降解需要200~500年。再加上使用量大、平均使用周期短(40%的塑料使用周期不到一年)和不恰当的后处置,已经造成了严重的环境污染和资源浪费。据统计,全球每年有480~1270 万吨的塑料流入大海,并随着洋流扩散到全球各地,有的甚至还会沉入海底。此外,有研究表明,塑料会在太阳光照射、生物降解和风化作用下逐渐降解为微型(<5mm)或纳米(<1000nm)颗粒,这些微塑料能吸附环境中的重金属、病原体和有机污染物,并最终通过食物链威胁到人类的生命健康。
我国作为发展中大国,积极承担国际责任,为积极应对全球塑料污染问题贡献中国智慧和中国方案。近十几年来我国陆续颁布了相关政策来应对塑料导致的“白色污染”。自2007年12月颁布《关于限制生产销售使用塑料购物袋》以来,国家鼓励使用生物可降解制品;2020 年以来,国家发改委又先后通过了《关于进一步加强塑料污染治理的意见》和被称为“史上最严禁塑令”的《关于扎实推进塑料污染治理工作的通知》等。这些政策在很大程度上促进了废弃塑料的回收和再利用。因此,无论从资源利用,还是从环境保护的角度出发,开展塑料的回收利用已是全社会的共识。
1 废塑料循环技术概述
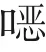
机械循环(mechanical recycling)主要涉及塑料的一级回收和二级回收。一级回收是指将未受污染的塑料进行简单回收利用。而在二级回收中,废弃塑料经过收集、分类、清洗、干燥、破碎、熔融、造粒等过程后被直接加工成适合市场需求的新塑料。但该方法对原料纯度要求较高,无法很好地处理混合或受污染的生活塑料垃圾。此外,物理回收过程中的机械、热等作用会导致再生塑料性能下降,不再适合制作高档次的塑料制品,其应用面受到一定的限制,因此多为降级回收。
化学循环(chemical recycling)是指通过化学手段将废弃塑料转化为小分子(通常为气体或液体)的技术,这些小分子可进一步转化为相关石化产品或塑料原料。该方法大致上可分为热裂解、催化热解和化学解聚。通过热裂解和催化热解得到热解气体(C~C)、液体油和蜡,通常可用作化学原料或低值燃料。化学解聚则通过不同的化学反应(如水解、醇解、糖酵解等)重新回收聚合物单体(闭环回收)或者将其转化为其他有用的化学品或材料(开环回收)。按使用的催化剂或溶剂的不同,化学解聚回收单体的技术则又可分为水解、醇解、氨解等。回收的聚合物单体在被纯化后可再聚合为新的塑料,这在理论上可以实现无限循环。热裂解和催化热解能够处理不同类型塑料的混合物,而化学解聚法多针对于某一特定类型的塑料。
在国家提出2030 年碳达峰、2060 年碳中和目标的大背景下,发展更加简单高效的回收方式,将废弃塑料高值化利用具有重要意义,循环经济也必将成为塑料行业未来的发展方向。2019 年,美国能源部首次提出化学升级再造(chemical upcycling)是未来解决废弃塑料污染的关键手段。化学升级再造是指通过发掘塑料中碳、氢、氧和大分子结构的内在价值,并将其转化为高品质化学品、燃料和功能型材料。相比较,化学升级再造可被视为未来唯一可实现可持续发展的废弃塑料高值化利用方法。相比于传统的机械循环和化学循环技术,升级再造策略不仅有助于显著提升产物的经济价值,降低转化过程的能源消耗和二次污染,还有望实现混合塑料的直接转化利用。
2 催化热解
2.1 催化热解制热解油
热裂解通常是在无氧或低氧环境、400~800℃高温下进行。反应过程涉及聚合物链的无序断裂和链端断裂,因而热解产物中烃类碳数分布宽,产物选择性差,产物中大量的烯烃和石蜡容易堵住管路等。近年来直接热解的相关研究较少,故本文不对其作过多介绍。
催化热解通常用于聚烯烃塑料,如聚乙烯、聚丙烯等。合适催化剂的存在可以显著降低反应的活化能,降低反应温度,提高塑料的转化率和液体油收率,催化热解产物分布也易于控制,提高了液体油的质量。塑料催化热解过程主要涉及自由基机理和碳正离子反应机理等引发的随机断链反应、异构化反应、脱氢反应、氢转移反应、芳构化反应等,具体反应机理受催化剂活性影响。常见的废塑料催化热解催化剂有各类沸石分子筛、活性炭、催化裂化催化剂、金属氧化物等。
2.1.1 分子筛催化剂
沸石分子筛是聚烯烃催化热解领域研究最早、应用最广的催化剂。分子筛的比表面积、孔隙结构和酸度对热解产物的选择性有显著影响。Bagri等使用Y型沸石和ZMS-5分子筛研究了催化剂孔隙对PE 催化热解的影响。当催化温度为600℃时,两种催化剂都获得了最高的芳烃产率,分别为37.2%和7.7%,其中Y型沸石热解油中的单环芳烃收率达到32.7%。Y 型沸石因其更大的孔径、更强的酸度因而具有较高催化芳构化的能力,而ZSM-5 的微孔结构有利于小分子轻烃的形成。Caldeira等通过向β沸石中加入介孔模板剂十六烷基三甲基溴化铵(CTAB)或者在β 沸石外部接枝硅烷化试剂制备了具有不同介孔尺寸分布的分级β 沸石。相比于传统方法合成的β沸石,两种分级β沸石在低密度聚乙烯(LDPE)催化热解中都展现了更好的催化性能。Aguado 等发现与HZSM-5 相比,Al-MCM-41 在450℃下具有较高的催化活性,LDPE 热解产物以高烯烃含量的可冷凝汽油馏分为主。这是由Al-MCM-41 具有的弱酸性和较大孔结构导致的。Ratnasari 等采用两段式固定床反应器考察了两种真实塑料垃圾(矿泉水塑料瓶和家用食品包装)在MCM-41 和ZSM-5 混合物作用下的催化热解行为。实验结果表明,当反应温度为500℃、MCM-41/ZSM-5 为1/1 时,两种废塑料热解油中芳烃质量分数分别高达95.14%和88.74%。研究认为具有较大比表面积和孔体积的MCM-41有利于大分子塑料的初步裂解,随后这些小分子扩散进较高酸度的ZSM-5微孔内进行重整,产生以C~C为主的芳烃。Elordi 等采用锥形喷射床反应器研究了高密度聚乙烯(HDPE) 在HZSM-5、HY 和Hβ 上500℃的热解反应。结果表明以HZSM-5 为催化剂能够获得最高的单环芳烃收率。他们认为HZSM-5较强的择形作用和高Brønsted/Lewis 酸比促进了大分子链的断裂并抑制了缩合和氢转移反应,而HY最能促进氢转移反应。
此外,产物收率和组成还与热解温度、载气流速、催化剂与塑料比有关。Xue 等以HZSM-5 为催化剂,研究了塑料类型、催化剂-塑料接触模式和载气类型对于热解油产物组分的影响。实验结果表明,在催化温度为600℃时,聚苯乙烯(PS)的芳烃收率最高,达到85%,而聚乙烯(PE)和聚丙烯(PP)的热解油则以烯烃和烷烃为主,芳烃的产率分别仅为26.5%和26.6%。对于聚对苯二甲酸乙二醇酯(PET)、PE和PP,原位催化热解的芳烃收率高于异位催化,PS 则刚好相反。此外,与惰性气体作为载气相比,以氢气作为载气显著降低了焦炭的产量并提高了单环芳烃的收率和选择性。研究认为,聚合物分子链首先在HZSM-5 表面的Lewis 酸性位发生氢抽提反应,形成碳正离子中间体,然后经过裂解、低聚和芳构化等过程形成芳烃。
在沸石分子筛上负载金属可以制备同时具有金属活性位点和酸性功能的双功能催化剂。金属活性位点可以促进脱氢/加氢反应的进行,而酸性位点可以催化芳构化反应。Renzini等将负载质量分数为2.84%Zn的ZSM-11(Zn-ZSM-11)和HZSM-11 分别用于LDPE 的催化热解。当催化温度为500℃、塑料与催化剂比为0.5时,液体油产率分别为55%和42%,且C~C芳烃占比分别为96%和63%。研究认为,HZSM-11上更多的Brønsted酸性位有利于断裂和五配位碳正离子的质子迁移活性,导致了更高的气体产率。而Zn-ZSM-11 对芳烃的高选择性是因为Zn的引入形成了大量的Lewis酸性位,促进了环化-氢转移反应。
相比于传统的固定床加热,微波加热通过物料能量耗散进而对介质直接加热,具有传热效率高、加热均匀、能耗低和选择性加热等优势。Zhang等以商用ZSM-5为催化剂,首次研究了微波裂解LDPE生产单环芳烃这一过程。实验结果表明,温度对芳烃产率和选择性发挥着重要作用。从249℃升至375℃后,单环芳烃产率从79.3%升高至84.34%,其中二甲苯占比超过50%,这归因于高温下催化剂的扩散系数降低,具有高扩散系数的二甲苯很容易从ZSM-5的孔隙中扩散出来。Ding等报道了一种微波热解与异位催化串联过程。首先,LDPE 在微波反应器中被NiO裂解和重整,生成烯烃。然后将微波反应器与装填有HY沸石的催化床串联,通过Diels-Alder反应将重整后的烯烃气体进一步转化为芳烃和异构烷烃。在热解温度为500℃、催化温度为450℃、催化剂与塑料的最佳比例为1∶10 条件下,液体油最大产率达到56.54%。Fan 等在此基础上将间歇式反应器升级为半连续进料,避免了因多次填料间隙造成的能量损失。较快的进料速度会导致更多焦炭和重烃组分的形成,这可能是因为HZSM-5上有限的催化位点无法在短时间内催化过多的热解气体。
2.1.2 活性炭催化剂
活性炭因其来源广泛、孔隙结构发达和高比表面积而在环境污染物降解、生物质和塑料催化等领域极具应用前景。然而其本身的催化性能较弱,无法满足特定反应的需要,因此科研人员通过引入各种活化方法来调节活性炭的表面化学性质,进而提高其催化性能。常见的活化方法有物理活化和化学活化。物理活化过程中水蒸气或CO等活化气体在高温下与活性炭内部不稳定的碳原子结合并以CO+H或者CO 的形式逸出,从而在活性炭内部形成发达的孔隙结构。所以活化时间、活化温度和活化气体流量等参数决定着活性炭的比表面积和孔隙率。化学活化则将酸(HPO、HNO、HO等)、碱(KOH、NaOH)或盐类(ZnCl、KCO)与活性炭按一定比例混合,再在惰性气氛中升温活化。活化剂的类型和浸渍比是影响活性炭性能的重要因素。酸、碱活化都有助于增大比表面积和孔隙率,但使用不同的酸活化剂可以在活性炭表面引入不同的官能团,而碱活化则会产生表面正电荷,吸附带负电的物质。
Zhang 等研究了不同活化方法对商用活性炭催化热解聚乙烯的性能影响。比表面积测试(BET)结果表明,水蒸气活化后的活性炭主要呈现微孔结构,且比表面积(891.12m/g)和孔容(0.52cm/g)都远小于磷酸活化的活性炭(比表面积1266.68m/g,孔容1.02cm/g)。此外,磷酸活化在活性炭表面引入了大量的含磷及含氧酸性官能团,提高了表面酸度(0.436mmol NH/g),而水蒸气活化的活性炭的酸度仅为0.012mmol NH/g。进一步的研究发现,改性活性炭上的弱酸和中强酸分别有利于烷烃和芳烃的形成,所获得的液体油几乎可完全用于航空燃油的生产,其中烷烃和芳烃分别占据71.3%和28.7%(图1)。
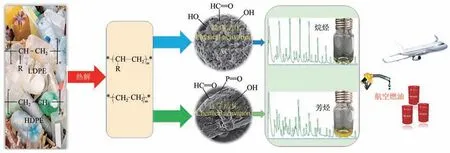
图1 磷酸改性活性炭催化热解聚乙烯制备航空燃油
Sun 等使用磷酸活化的活性炭催化聚乙烯芳构化。当磷酸浸渍比较低时,未形成明显的孔结构。当浸渍比超过50%时,过强的活化作用降低了孔壁的机械强度,造成孔的塌陷。此外高浓度的磷酸在活化过程中形成了大量的PO造成孔道的堵塞。当浸渍比为40%时,比表面积(742.9m/g)和孔容(0.86cm/g)达到峰值,保证了最高的传质速率。在浸渍比为40%,停留时间为3s 的最优操作条件下,PE 热解油中的芳烃含量达到30%,其中单环芳烃占比约79.3%。研究表明,磷酸活化后引入的Brønsted 酸性位,如C—O—PO、C—PO、及C—PO中的P—OH 催化的氢转移反应,以及P==O 等脱氢活性位催化的直接脱氢反应是芳构化的两种主要途径。
Duan 等系统研究了HPO浓度、活性炭炭化温度、催化温度和活性炭与LDPE的比例对热解油收率和产物分布的影响。提高HPO浓度和炭化温度可以增加比表面积、孔体积和酸性位点,但是结焦也会更严重。当催化温度从450℃升高至600℃时,焦炭产率明显下降。
相较于HPO活化引入Brønsted 酸性位,ZnCl活化则可以在活性炭表面引入锌类活性物质Zn(Ⅱ)作为Lewis 酸性位。这些Lewis 酸性位可以通过Diels-Alder、氢转移、直接环化脱氢等反应将烯烃转化为芳烃。此外,Lewis 酸性位可以促进芳烃的烷基化,促进轻烃的进一步转化和更多支链芳烃的生成。
2.1.3 其他类型催化剂
除沸石分子筛和活性炭外,少量研究采用催化裂化(FCC)催化剂、黏土和金属氧化物等材料作为催化剂。
FCC催化剂是一种通过黏结剂将活性组分(一般为沸石,如Y型和ZSM-5)和基质(陶土、氧化铝、氧化硅等)黏结在一起而组成的多组分催化剂,具有大孔径和高活性等特点,因此被广泛应用于重油轻质化制备柴油、汽油等高附加值产品。鉴于塑料也属于长链烷烃,则FCC 催化剂理论上也能够用来催化裂解塑料。Lee 等采用搅拌式半间歇性反应器在400℃、塑料与催化剂比为10∶1 的条件下研究了废FCC 催化剂对PS 和HDPE 混合物的催化芳构化反应,苯乙烯和乙苯是主要的芳烃产物。反应初期热解油的主要成分是轻质烃类,其中C~C组分占比超过90%。芳烃产率随着混合物中PS 占比提高而增加,但随着反应时间的增加而减少。研究认为,PS 和HDPE 的协同作用加强了PS热解产物的二次反应,导致了主要芳烃产物含量的下降。Vollmer 等发现相较于新制备的FCC 催化剂,炼油厂废弃的FCC 催化剂表现出更好的催化效果,这归因于基底部分在长期反应过程中沉积了Fe、Ni、V等少量金属颗粒,而这些金属颗粒有利于热裂解聚丙烯,并催化裂解产物芳香化。此外,红外光谱和共焦荧光显微镜证明,催化剂基体中的陶土和氧化铝等具有裂解-芳构化等功能,而沸石区域则会诱发积炭的形成。
Manos 等比较了HDPE 在黏土和USY 沸石催化作用下的热裂解特性,结果表明,当反应温度为600K时,HDPE在黏土催化作用下可以得到更高的液体产物(约70%),而在相同操作条件下,USY沸石仅能得到约50%液态产物。这归因于黏土催化剂酸性较弱,一方面可以防止催化热解反应中的过度裂解,有效降低氢转移反应,生成更多的液态烯烃;另一方面可显著降低焦炭的产生。Li等将Fe、Al、Ti 和Zr 的氧化物嵌入到具有层间结构的柱状黏土中并应用于混合塑料的催化热解中。结果表明,以Fe修饰的黏土作为催化剂可以获得最高的热解油产率和芳烃含量,分别为79.3%和44.5%。Fe修饰的黏土表面的Brønsted酸性位主要来自于蒙脱土结构中的羟基,而Lewis 酸性位则来自于金属氧化物。Brønsted 酸性位和Lewis 酸性位的协同作用是重质芳烃形成的主要原因。
碱金属氧化物常用于催化热解聚苯乙烯选择性制备苯乙烯单体。与固体酸催化机理不同,碱金属氧化物上的活性碱性位点首先会进攻碳链上的氢原子,形成碳负离子,然后发生断裂生成苯乙烯。Zhang 等研究了聚苯乙烯分别在固体酸和固体碱催化作用下的热解行为,发现碱金属氧化物催化裂解聚苯乙烯生产苯乙烯效果比固体酸催化剂好,其中BaO 表现出最佳催化活性。在350℃、塑料与催化剂比为10∶1的条件下,得到的液体油和苯乙烯收率最高,分别为93.4%和76.4%。
2.2 催化热解制碳纳米管
废塑料催化热解除了能获得燃料油,还可通过催化裂解-重整的方法获得高附加值的碳纳米管(carbon nanotubes,CNTs)和富氢气体。这一过程涉及气态烃类在催化剂表面吸附并裂解成碳自由基,然后碳自由基在催化剂表面扩散和自组装生长出CNTs(图2)。这不仅有利于降低CNTs 的生产成本,还可实现废弃塑料的资源化利用。催化剂在CNTs 的形成过程中起着关键作用,一方面可以促进长链塑料断键形成小分子气体烃类,另一方面能够降低催化裂解和重整反应所需要的温度,缩短反应时间。
图2 热解-重整法和微波激发法制备碳纳米管和氢气
催化剂一般由活性金属组分和载体构成。活性金属组分一般由一种或多种金属组成,这些活性金属既是塑料裂解的催化活性中心,又是CNTs 的生长位点。载体则可以改善活性金属的分散性,提高催化剂的活性和稳定性。相较于单金属催化剂,多金属催化剂比单金属催化剂表现出更好的活性和稳定性,且金属在催化过程中各自承担着不同的作用。Wu等使用共沉淀法制备了Ni-Mn-Al催化剂并研究了其催化热解聚丙烯制备碳纳米管。研究人员认为,Ni 金属颗粒的存在是碳纳米管形成和生长的关键条件,而Mn的存在可以提高碳纳米管的产量。Yao 等研究了Ni-Fe/γ-AlO双金属催化剂热解混合废塑料制备氢气和碳纳米管。改变双金属催化剂中Ni 和Fe 的相对含量发现,Ni 含量的增加有利于增强金属活性组分与载体间的相互作用,并且有助于提高碳纳米管的纯度和石墨化程度。而Fe 相对含量的增加可以明显提升H和固体碳的产率。
除了金属活性组分外,载体的选择对碳纳米管的品质也有影响。因为载体与金属之间的相互作用可以分散金属活性组分,避免金属颗粒的团聚,提高催化剂的活性和稳定性。一般来说载体表面的碱性位点越多,相互作用越强;酸性位点越多,相互作用越弱。Yao 等发现α-AlO载体由于载体-金属相互作用较弱导致金属活性组分发生团聚,催化剂颗粒粒径较大,而γ-AlO发达的孔结构可以更好地分散金属颗粒。而催化剂颗粒的大小决定着碳纳米管的直径。碳纳米管主要是以顶部生长模型形成,金属颗粒均匀地分散在碳纳米管的中间或顶部,以α-AlO为载体负载的催化剂得到的碳纳米管的直径比γ-AlO对应的碳纳米管直径大,且碳纳米管纯度较低。Jia 等使用原位生长的方法将Ni负载在金属氧化物上,并探究了Ni/Mg、Ni/Sr和Ni/La三种催化剂中金属-载体相互作用对催化热解塑料生成碳纳米管的影响。研究发现Ni/Mg催化剂中过强的金属-载体相互作用不利于Ni 颗粒的出溶,Ni/Sr中太弱的金属-载体作用限制了Ni在载体上的均匀分布,而Ni/La 适当强度的金属-载体相互作用对载体上Ni 颗粒的尺寸、分布和表面覆盖率起着重要作用。Aboul-Enein 等使用湿法浸渍的方法制备了Ni-Cu/LaO催化剂,并用于聚丙烯催化热解制备CNTs。研究发现Ni-Cu 与LaO之间良好的相互作用促进了金属颗粒的均匀分散,有效抑制了Ni-Cu 在高温下的团聚,从而改善了催化性能。
传统的热解-重整两步法需要较大的能量投入,且会产生大量的CO(每生产1kg H就会产生12kg CO)。Edwards 等将硝酸铁、硝酸铝和柠檬酸等摩尔混合得到的橙色凝胶在350℃下煅烧3h,制备了一种微波敏感催化剂(FeAlO)。在微波的激发作用下,FeAlO可在30~90 s内将废弃塑料一步转化为氢气和CNTs(图2)。连续十次循环之后,碳的总产率为620mg/g,其中CNTs的质量分数约为92%。此外,每克塑料的氢气产率高达55.6mmol(理论最高为71.4mmol)。这种微波激活法之所以获得了较高的氢气产率和纯度较高的CNTs,一方面是因为微波加热极大地减少了常规塑料热解中的副反应,使得C—H的断裂占主导地位;另一方面剩下的碳渗入Fe 内部形成FeC,在表面张力和轴向压力的作用下转化为CNTs。
3 化学解聚
与聚烯烃相比,通过C—O 键连接的PET 的解聚反应通常具有相对较低的反应能垒,这是因为C—O 键通常比C—C 和C—H 键更不稳定。这类反应通常使用亲核试剂来进攻羰基从而解聚塑料得到聚合物单体。催化剂的作用在于加快反应速率和提高产物的选择性。
借助酯基的水解,PET能够在碱性、中性和酸性条件下水解生成对苯二甲酸(terephthalic acid,TPA)和乙二醇(ethylene glycol,EG)。TPA和EG又是生产原生PET的原料,从而实现PET的闭环回收利用。碱性水解通常在质量分数为4%~20%的NaOH 或KOH 的水溶液中进行,在200~250℃温度和1.4~2.0MPa 压力条件下反应数小时,生成TPA的二钠盐或二钾盐,然后通过酸化沉淀得到TPA。中性水解一般在1~4MPa、200~300℃温度的水或水蒸气中进行。酸性水解通常以浓硫酸、硝酸或磷酸等无机酸为催化剂,反应条件相对温和(常压且温度一般不高于100℃)。因此水解法通常需要较高的温度和压力,能耗较大,且强酸强碱条件容易造成设备腐蚀。此外,水解产物杂质较多,目标产物TPA的分离提纯工艺相对复杂。
醇解法是目前工业应用最成熟的解聚方法。根据醇解剂的不同,醇解法一般可分为一元醇解法和二元醇解法。一元醇解法的优势在于醇解产物对苯二甲酸二甲酯(dimethyl terephthalate,DMT)的纯度较高。但反应条件苛刻、已造成设备腐蚀、产物分离成本较高。目前二元醇解法是最具吸引力的方法。二元醇解法的优势在于反应条件温和,醇解产物对苯二甲酸双羟乙酯[bis(hydroxyethyl)terephthalate,BHET]可用于再生产PET,聚氨酯和塑料助剂。二元醇解法通常是在180~240℃温度范围内进行,常用的醇解剂包括乙二醇(ethylene glycol,EG)、丙二醇(propylene glycol,PG)、1,4-丁二醇(1,4-butanediol,BDO)等,其中以乙二醇的应用最为广泛和成熟。反应在酯交换催化剂的作用下,PET 酯键断裂并被羟基取代,完全解聚为BHET单体。本节主要以二元醇解法为例,总结了离子液体(ionic liquids,ILs)和深共晶溶剂(deep eutectic solvents,DESs)在PET 化学分解领域的最新研究进展。表1总结了使用不同催化剂乙二醇解PET的最佳条件。
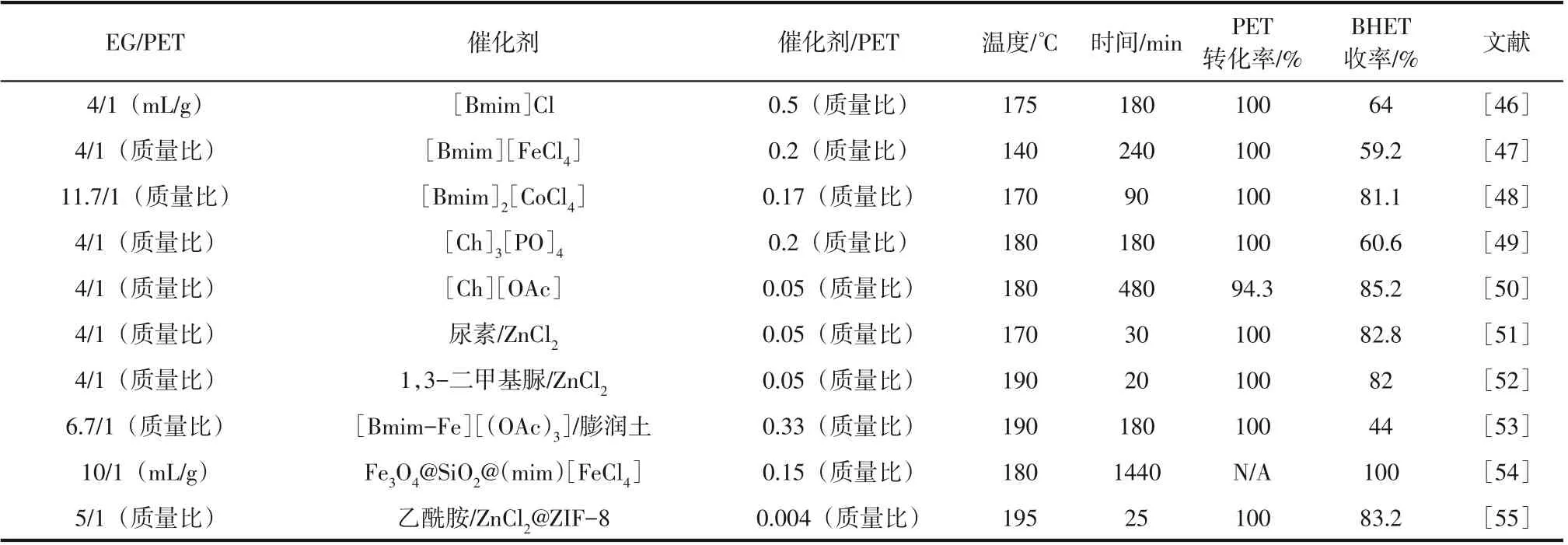
表1 不同催化剂催化醇解PET的性能对比
3.1 离子液体
离子液体具有不易挥发、热稳定性好的特点,且可通过广泛选择阴阳离子组合来优化自身性质。此外。相比于传统催化剂,离子液体能够很好地溶解于EG,更容易与PET接触反应,催化效率更高。2009年,Wang等首次报道了离子液体催化PET降解,并研究了其酸碱性对于催化性能的影响。研究发现,酸性离子液体在温度高于180℃时不能稳定存在。虽然碱性离子液体表现出最强的催化活性,但价格昂贵且制备复杂。相比较而言,中性离子液体([Bmim]Cl 和[Bmim]Br])才是最佳选择。180℃、5g PET、20g EG、反应8h,PET 的转化率分别为44.7%和98.7%,但是1-丁基-3-甲基咪唑氯盐([Bmim]Cl)的用量较大。Alnaqbi等探究了微波辅助[Bmim]Cl 催化降解PET,微波辐射加热,175℃、5g PET、20mL EG、2.5g 催化剂、反应3h,PET 转化率和BHET 收率分别为100%和64%。相较于传统加热方式,在达到相同的转化率和收率前提下,微波辅助加热可节约6~8h。
有研究发现,相较于不含金属的离子液体,含金属的离子液体在PET降解过程中表现出更好的催化性能,这可能是因为离子液体中的阴、阳离子存在着协同催化作用。Wang等将[Bmim]Cl和FeCl等摩尔混合,制备了Lewis酸性离子液体[Bmim][FeCl],在常压、140℃、5g PET、1g催化剂、20g EG条件下反应4h,PET 转化率和BHET 收率分别为100%和59.2%。但铁离子的存在使得产品带有颜色,水洗等提纯步骤造成产物流失,收率下降。机理表明,[Bmim]与PET酯基上的氧相互作用,然后EG的羟基氧进攻酯基上的碳正离子,形成四面体中间产物。EG 上的羟基氢在[FeCl]的作用下脱离,酰氧键断裂—OCHCH—与H结合形成—OCHCHH。该过程反复进行,最后得到单体BHET[图3(a)]。Wang 等将一系列过渡金属氯盐(MnCl、CoCl、NiCl、CuCl、ZnCl、FeCl和CrCl)分别与[Bmim]Cl 混合,发现[Bmim][CoCl]表现出最佳的催化性能,常压、170℃、5g PET、20g EG、1g 催化剂条件下反应4h,PET转化率和BHET收率分别为100%和81.1%。
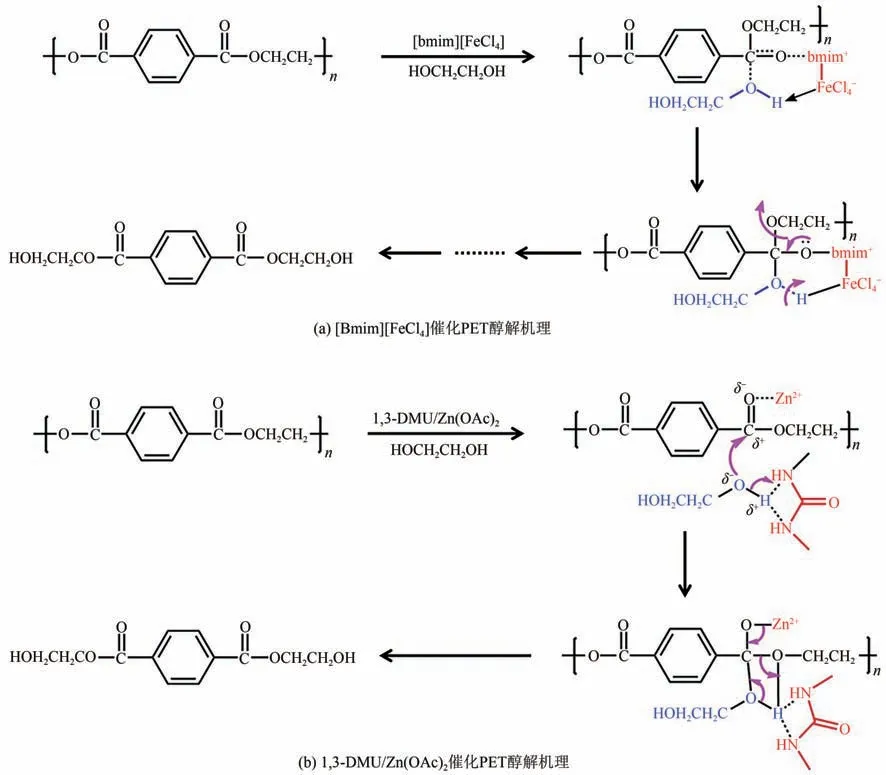
图3 不同催化剂催化PET醇解机理示意图
传统金属咪唑类离子液体虽然具有较高的催化活性,但是金属极易残留于产物中,且咪唑对微生物也具有毒性。针对这一问题,Sun 等合成了一种具有生物相容性的胆碱磷酸盐([Ch][PO]),180℃、0.5g PET、2g EG、0.1g 催化剂条件下反应4h,PET 转化率和BHET 收率分别为100% 和60.6%。最近,Liu等合成了一系列不含金属的胆碱类离子液体,经过筛选发现由氢氧化胆碱和醋酸或甲酸合成的催化剂([Ch][For]、[Ch][OAc])具有较强的催化活性。在常压、180℃、5g PET、20g EG、质量分数为5%的催化剂条件下反应8h,PET 转化率和BHET 收率分别为94.3%和85.2%,几乎可以和金属基离子液体的催化效果相媲美。密度泛函理论计算(DFT)结果表明,[OAc]上的氧与EG中的羟基氢形成氢键,增加了EG 中羟基氧的电负性,增强了EG 的亲核进攻能力。同时,胆碱阳离子上的羟基氢质子化PET 中的羰基碳,使其更具亲电子性。
3.2 深共晶溶剂
深共晶溶剂因其成本低、制备简单、毒性低、生物相容性好并与离子液体具有类似的特性,因而也被广泛用作PET 的二元醇解剂。Wang 等设计了一种尿素/ZnCl[(尿素)/(ZnCl)=4]深共晶溶剂,在170℃下反应30min,PET的转化率和BHET的收率分别为100%和83%。DFT计算表明,EG的羟基氢与尿素上的羰基氧形成氢键,增加了EG 中羟基O—H 的键长,使EG 中的羟基氧具有更强的电负性,因此EG 中的羟基氧更容易亲核进攻PET 酯基上的羰基。同时,Zn与PET 上的氧产生配位作用,在氢键与配位键的协同作用下PET、EG与Zn之间形成环状过渡态进而使PET链断开。
Liu 等设计了一种1,3-二甲基脲/醋酸锌[1,3-DMU/Zn(OAc)]深共晶溶剂,实现了PET 的高效降解。190℃温度下反应20min 就实现了PET 的完全转化,BHET 的收率达到82%。1,3-DMU 的活性中心是氨基,而Zn(OAc)则表现出Lewis 酸催化活性,1,3-DMU和Zn(OAc)之间的酸碱协同催化作用是其催化性能优异的根本原因。核磁共振氢谱(H NMR)证明了1,3-DMU 的氨基与EG 的羟基H存在着强烈的氢键作用,从而增强了EG 上羟基O的电负性,更易于进攻PET 酯基上的羰基。同时,Zn作为路易斯酸能够质子化PET 酯基上的羰基,使羰基上的C具有更强的亲电子性,因此更易受到EG的亲核进攻[图3(b)]。
尽管离子液体和深共晶溶剂在催化活性和选择性上极具优势,但是要将这两种液体催化剂从乙二醇中分离出来也是极其困难的。一般可通过减压蒸发溶剂的方法来实现回收利用,但会消耗大量的能量。为了解决液体催化剂回收困难的问题,Al-Sabagh 等采用湿法浸渍法将[Bmim-Fe][(OAc)]负载在膨润土上,在190℃、3g PET、20g EG、1g 催化剂条件下反应3h,PET转化率达到100%,BHET单体的收率为44%。经过6次循环未见任何催化剂活性下降。Cano等首次报道了使用顺磁性含铁离子液体对SiO包覆的磁性FeO进行表面改性,制备了一种可通过磁性回收的高活性纳米催化剂FeO@SiO@(mim)[FeCl],用于PET的解聚,BHET的收率和选择性接近100%。最重要的是,在外部磁场作用下可以在3min 内回收催化剂。催化剂连续循环12 次,催化活性没有明显的降低,表现出良好的循环性能。Wang 等使用乙酰胺和氯化锌合成了DESs,并将其负载在沸石咪唑酯骨架ZIF-8上,合成了一种新型DES@ZIF-8催化剂,195℃下反应25min,PET 转化率和BHET 收率分别为100%和83.2%。只需简单过滤即可实现催化剂的回收。经过6次的循环使用催化剂活性没有明显降低。
4 催化氢解
塑料催化氢解(hydrogenolysis)一般是指在催化剂和氢气作用下,聚合物骨架中碳碳键断裂,生成短链饱和烷烃的过程。目前催化氢解主要用于处理PE 和PET。表2 总结了不同催化剂催化氢解废塑料的性能对比。
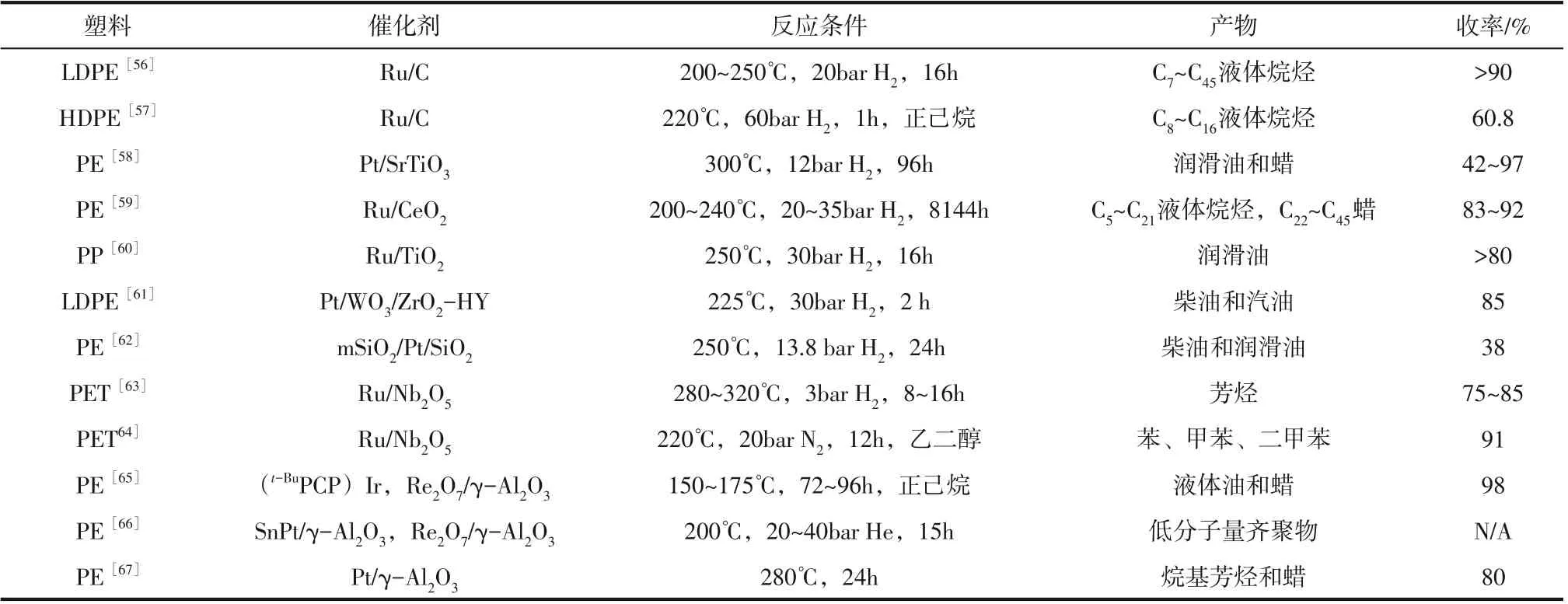
表2 不同催化剂催化氢解废塑料的最佳操作条件及产物对比
加氢活性金属(或金属氧化物)负载催化剂是废塑料氢解最常用的催化剂。其中,炭因其较高的热稳定性而成为最常用的载体。Rorrer 等首先用十八烷作为PE 模型化合物筛选了一系列贵金属催化剂,发现Ru/C催化剂在200~250℃温度范围内具有最高的催化活性。反应温度为200℃时,气体产物主要为甲烷、乙烷、丙烷和丁烷,但随着温度升高至250℃,气体产物中只剩下甲烷。随着H压力的增大,固体烷烃(>C)逐渐转化为液态烃类(C~C),最终变成气态轻烃(C~C)。之后,研究人员发现,纯LDPE 和消费后LDPE 塑料瓶也可在200℃、20bar(1bar=10Pa)H、反应16h 条件下转化为C~C的烃类,证明了Ru/C 催化剂的普适性。Lin等探究了溶剂效应和氢气压力对于Ru/C催化氢解塑料体系的影响。塑料在不同溶剂中的溶解度不同,解聚反应发生的机制也不尽相同,发现生产最多燃料的溶剂是正己烷,而甲基环己烷则是生产高质量润滑剂的最佳选择[图4(a)]。在较低氢气压力下,HDPE主要是以链端断裂为主,随着氢气压力的增大,内部断键逐渐占主导地位[图4(b)]。喷气燃料和润滑剂类型碳氢化合物的最大产率分别为60.8%和31.6%。优化反应条件后,液态烃类产品的总产率在220℃、1 h内就达到了约90%。
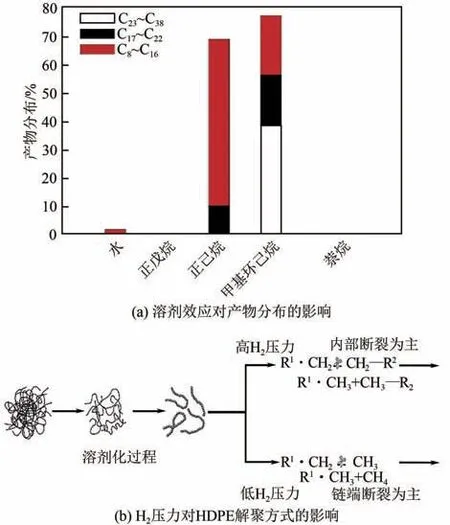
图4 溶剂效应和H2压力对产物分布和断键方式的影响
除炭载体之外,加氢活性金属还被负载在多种金属氧化物上。这种金属-载体的相互作用对于调控催化剂性能也起着重要作用。Celik 等采用原子层沉积的方法,在SrTiO纳米立方体上负载了高度分散的Pt 纳米颗粒(Pt/SrTiO)。在300℃、1.38MPa H压力条件下反应96h,不同分子量的聚乙烯和市售的塑料袋可被转化为高品质的润滑油(200~1000Da)和蜡,产率为42%~99%。DFT计算结果表明,聚乙烯链更倾向于吸附在Pt 表面。同时,Pt纳米颗粒表面更多的低配位点抑制了过度氢解,避免了轻质烃的生成。此外,Pt 纳米颗粒与SrTiO之间强烈的金属-载体相互作用能显著缓解催化剂的烧结。Nakaji 等使用Ru/CeO催化剂在200℃、20bar H条件下将LDPE 氢解为液态烃类(C~C)和蜡(C~C),总收率达到92%。研究人员推测Ru/CeO出色的催化性能归因于LDPE 在纳米Ru 金属上较低的C—C 断键自由。Liu 等使用湿法浸渍法,以TiO为载体负载了一系列金属(Pd、Rh、Ir、Ni、Pt、Ru),发现Ru/TiO对聚丙烯转化为液体产品的活性最高。研究发现,通过调控催化剂的添加量可以有效调控产物分布。在催化剂添加量较高时候,产物的分子量下降较快。核磁和红外光谱结果表明,聚丙烯在Ru/TiO表面发生动态的吸脱附和内部C—C 键断裂。反应初期聚丙烯分子量较高时,自身结构立体规整性的打破是发生氢解反应的先决条件。此外,聚丙烯碳链中脱甲基反应减少了叔碳的数量,从而抑制液体产物进一步发生C—C裂解为甲烷和乙烷。
另一方面,双功能催化剂的引入也是设计高效催化剂的方法。Liu 等使用Pt/WO/ZrO和HY 沸石的混合物作为催化剂,在225℃下通过接力催化对塑料进行氢解,得到了由汽油、柴油和煤油组成的液态燃料产品,产率高达85%。研究发现单独使用Pt/WO/ZrO作为催化剂时,即便是在更高温度下,LDPE 的转化率也比较低,但是将Pt/WO/ZrO和HY沸石的混合物作为催化剂时,2h就可转化所有LDPE,说明两种催化材料之间存在极强的协同效应。采用吡啶对酸性位点进行毒化,发现塑料在Pt表面活化之后,活性中间体既可以在HY沸石的酸性位点上发生反应,也可以在WO/ZrO表面的酸性位点发生反应,最后在Pt 表面加氢获得饱和异构烷烃产物。另外,HY 沸石的酸性对转化过程也有重要影响。降低铝含量导致汽油产率从72%下降到32%,柴油质量分数从11%增加到27%。同时,降低酸密度会导致固体残余物的产量增加。此外,调控HY 沸石的孔道结构或改变固体酸种类,还可选择性调控液体产物分布为汽油,喷气燃料和柴油。
受自然界大分子解构酶将柔性大分子行进式链裁剪成原子级别片段的启发,Huang 等设计了一种基于介孔二氧化硅和铂纳米粒子的多级结构催化剂,在300℃、24h 和1.72MPa H压力条件下,将高密度聚乙烯“剪”成碳数分布集中的小分子烷烃片段,所得产物可用于柴油和润滑油的制备。这种新型催化剂呈现一种“有序介孔壳/活性位点/核”结构,铂纳米粒子作为催化活性位点被负载在二氧化硅壳层中介孔的底部(mSiO/Pt/SiO)。C 核磁共振谱图表明,聚乙烯链在商用硅胶上存在反式、移动和偏转三种构象,而在mSiO上只有反式和移动构象,说明介孔mSiO能诱导聚乙烯链形成长锯齿状结构[图5(a)]。在催化温度下,高密度聚乙烯长链可容易地在孔内移动,且进入孔内的链长与孔的长度相匹配,不过随后的逸出却受到聚合物-表面相互作用的抑制。之后,介孔底部的铂纳米粒子对孔内吸附的聚乙烯长链进行氢解反应。二氧化硅孔隙和长链聚合物之间的大量累积分散相互作用导致了强结合,而与小分子片段相互作用较小,导致小分子量的片段被不断释放,而聚合物则进一步穿入催化孔内进行定位并进行下一步的裁剪。这些步骤重复进行直至将整个聚合物链转化为所需的小分子量的烷烃片段[图5(b)]。有趣的是,催化剂孔径的大小会影响氢解产物的分布。通过调节催化剂孔径(1.7nm、2.4nm和3.5nm),可以分别得到以C、C和C为碳链分布中心的链烃混合物,为进一步转化和制造柴油以及润滑油提供了可能。
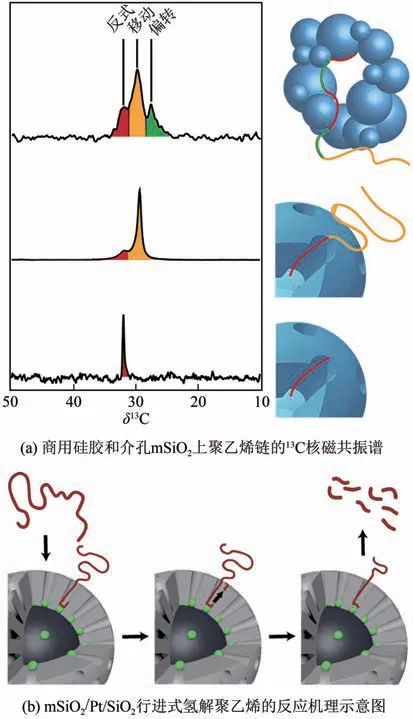
图5 商用硅胶和介孔mSiO2上聚乙烯的13C核磁共振谱图及mSiO2/Pt/SiO2行进式氢解聚乙烯的反应机理
Wang 等从化学键活化断裂的角度,采用双功能Ru/NbO催化剂,首次实现了废旧芳香塑料内C—O 或/和C—C 连接键的选择性断裂制备芳烃。相比于其他Ru催化剂,Ru/NbO中Ru的配位数仅为5~6,以配位不饱和的边缘位点为主。高度分散的Ru 纳米颗粒抑制了苯环和氢气的共吸附,避免了苯环的过度加氢,从而实现了较高的芳烃选择性[图6(a)]。同时,NbO上强的亲氧位点NbO具有优异的活化C—O 键和吸附苯环的能力,而其Brønsted 酸性位点则能质子化吸附的苯环,协助对C—C键的活化。在小颗粒Ru解离的氢物种的协同作用下,实现了对芳香塑料内C—O和C—C键的精准活化断裂。更具现实意义的是,这种Ru/NbO双功能催化剂不仅能实现单组分芳香塑料直接转化为芳烃,还能实现混合芳香塑料高选择性地制备芳烃化学品,芳烃收率高达75%~85%,且对二甲苯的选择性最高[图6(b)]。之后,他们发现了一条新的途径,通过使用乙二醇作为氢源、Ru/NbO作为催化剂,在220℃、2MPa N条件下反应12h,成功将PET 转化为苯、甲苯和二甲苯(BTX)。整个催化过程涉及三个串联步骤:首先是PET 先在NbO表面水解,然后Ru/NbO催化重整乙二醇原位产生氢气作为氢源,最后在Ru的催化下发生氢解/脱羧反应。XPS 结果表明,NbO表面高度分散着带正电荷的Ru 物种,说明Ru 和NbO载体之间存在较强的相互作用,从而有效地抑制了脱羧反应的发生。
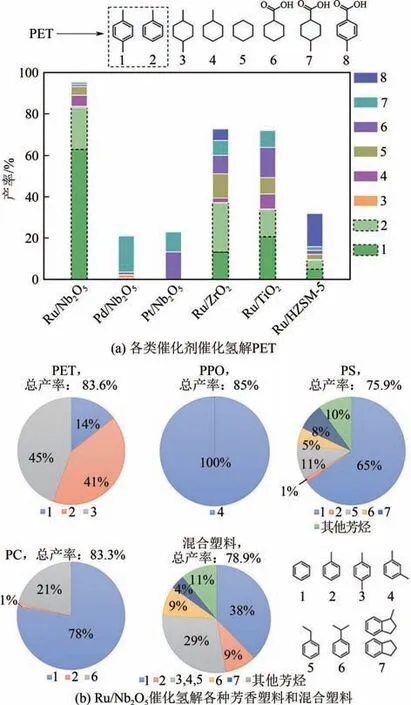
图6 各类金属负载催化剂催化氢解PET及Ru/Nb2O5催化氢解各种芳香塑料和混合塑料
除了以氢气分子和生物质作为外加氢源外,聚乙烯骨架中丰富的氢同样可以参与原位氢解反应。Huang 等报道了一种交叉烷烃复分解催化策略(tandem catalytic cross alkane metathesis)(图7),首先利用“钳形”Ir 配合物催化PE 和低碳烷烃发生脱氢反应,生成不饱和烯烃和Ir-H。然后,在ReO/AlO催化剂的作用下发生烯烃歧化反应,生成两个新的烯烃。最后,新生成的烯烃与Ir-H发生加氢反应得到饱和烷烃。体系中过量的低碳烷烃不断与PE发生重组反应,有效降低了PE的分子量和碳链长度,直至把分子量上万、甚至上百万的聚乙烯降解为可作为清洁柴油的烷烃。但是反应后剩余的过量低碳烷烃能否被有效回收利用尚未可知,且均相Ir 配合物催化剂的回收过程仍然十分棘手。针对这一问题,Beckham 等设计了一种非均相体系,将SnPt 和ReO均负载在γ-AlO上(SnPt/γ-AlO和ReO/γ-AlO),分别负责脱氢/加氢和烯烃歧化反应。以正戊烷为溶剂和低碳烷烃反应物,在200℃下反应15h,PE 的分子量下降了73%。但是,这两项工作使用了大量的Ir 和Re 等贵金属。因此需要进一步的努力来降低催化剂的用量,探究此类催化剂的再生和回收问题。当然最重要的还是探索是否有可替代Ir 和Re 的廉价金属,从而最大限度地降低成本。
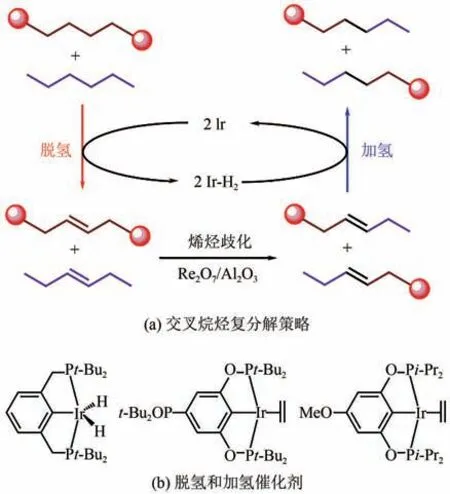
图7 聚乙烯交叉烷烃复分解制备低碳烷烃的反应机理及所使用的催化剂
此外,Scott等创新性地将放热氢解和吸热芳构化反应耦合起来,将芳构化释放的氢直接用于氢解低密度聚乙烯转化为长链烷基芳烃的混合物,提出了一种不需要溶剂和添加氢,在低温下将废弃的聚乙烯一步转化为更高价值的长链烷基芳烃的创新方法。只使用Pt/γ-AlO催化剂,在280℃下反应24h,液体产物和蜡的产率达到80%,烷基芳烃的选择性约为57%。该工作的难点在于氢解和芳构化两个反应之间必须保持微妙的平衡,因为一方面更高的温度会产生甲烷、乙烷和丙烷等价值较低的气体,以及可能导致催化剂失活的焦炭沉积物;另一方面,反应体系中氢的分压必须足够高以确保聚合物链被不断切割,但又足够必须低以避免所形成的有价值的烷基芳族化合物被过度氢化。
5 光催化
近年来,光催化重整(photoreforming)技术也被应用于废弃塑料的化学增值再造。塑料在合适的光催化剂作用下可以被转化为高附加值的H及其他小分子化学品,反应机理如图8所示。光催化剂在光照下产生光生电子和空穴,塑料作为电子给体消耗空穴被氧化为小分子化合物,而光生电子则从光催化剂的导带迁移到助催化剂上,将水还原为H。光催化重整技术利用取之不尽的太阳光作为能源,相比于其他热催化技术反应条件更加温和,可在常温常压下进行。
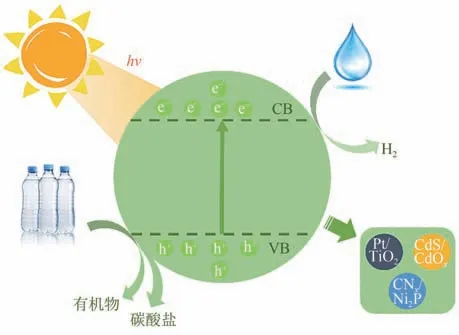
图8 废塑料光催化重整机理
这种“塑料到氢气”的概念最早可追溯到1981 年,Kawai 等使用铂负载的TiO(Pt/TiO)作为催化剂,在室温下以500W 氙灯为光源、PVC作为空穴捕获剂,成功实现了光解水制氢。然而Pt/TiO仅能吸收紫外光,催化剂活性低且成本较高,氢气的生成速率仅为1.3μmol/h。虽然当时并未提出确切的反应机理,但这项开创性的研究工作首次实现了废塑料的光催化制氢。
2018 年,Reisner 等以CdS/CdO量子点作为光催化剂,实现了可见光条件下PLA、PET 和PU的光催化重整制氢。为进一步提高反应效率,研究人员预先将塑料粉末在10mol/L NaOH 溶液中预处理24 h。相较于未预处理的塑料,在相同的反应条件下,H产率提升了4 倍。这是因为碱性条件下PET会水解为单体进而更容易地被光催化重整。研究人员使用现实中的PET 塑料瓶来验证CdS/CdO的普适性(图9),反应连续进行6 天,活性高达(4.13±0.40)mmol/(g·h),外部量子产率为2.17%±0.38%,转换率为5.15%±0.72%。虽然该体系规避了贵金属,但需要使用有毒的CdS。
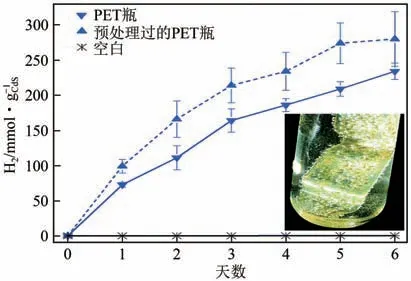
图9 CdS/CdOx光催化重整PET塑料瓶制备H2
为了解决镉的毒性问题,研究人员设计合成了一种廉价且无毒的氰胺功能化氮化碳与磷化镍(CN/NiP)复合高性能光催化剂。研究结果表明,氰胺改性使得氮化碳的带隙降低至2.7eV,将吸收光谱拓展到可见光区[图10(a)]。助催化剂NiP能有效促进CN光生载流子的分离和传输[图10(b)],同时充当反应活性位点。在碱性水溶液中,PET和PLA 成功被转化为清洁燃料H和多种有机化学品,包括醋酸盐、甲酸盐和乙二醛等。在10mol/L KOH溶液中,CN/NiP对PET的最大产氢量为111μmol/g;对于PLA,CN/NiP 的最大产氢量为211μmol/g[图10(c)]。使用KOH水溶液作为溶剂一方面可以增加塑料的溶解性,另一方面,NiP在碱性条件下表面可以形成一层Ni(OH),促进水的解离,提高析氢活性。使用对苯二甲酸(TPA)捕获·OH,反应后却并未检测到TPA-OH的信号,说明CN不足以氧化水生成·OH,研究认为这是一个以空穴为主导的氧化反应。CN/NiP 不仅能降解纯度较高的塑料,对于一些纤维,PET塑料瓶和油污染的PET材料也同样适用,在长达5天的循环实验中依然保持稳定的产氢性能。但以上两个光催化体系都需要在碱性环境下运行,所以后续废碱液的处理是一个严峻的问题。
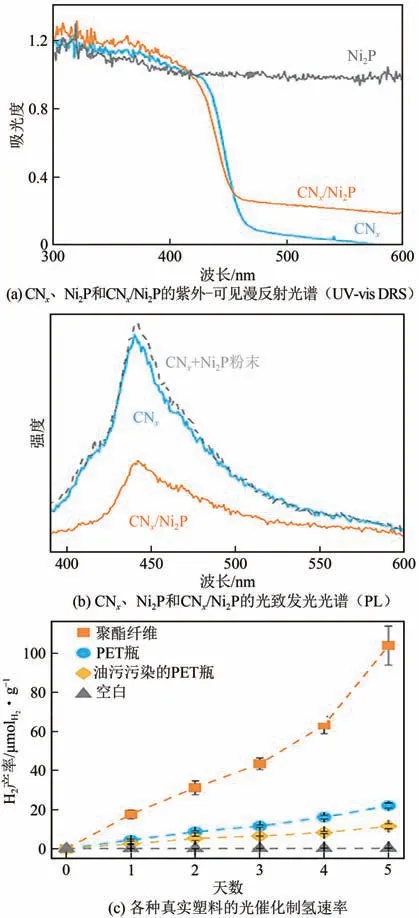
图10 CNx、Ni2P和CNx/Ni2P的UV-vis DRS和PL以及各种真实塑料的光催化产氢速率
除了H之外,塑料还可以被光催化转化为高品质的C化学品。Xie 等制备了单原子厚度的片状NbO,在模拟太阳光照射下,成功将一次性塑料袋(主要为聚乙烯)、餐盒(主要为聚丙烯)、保鲜膜(主要为聚氯乙烯)等塑料垃圾在水中转化为高能量密度的C化学品,且不需要加入牺牲剂。研究人员发现纯PE、PP和PVC可分别在40h、60h和90h 内被完全光降解为CO。随着反应的继续进行,CHCOOH 的生成速率逐渐增加。为了确认CO的来源,研究人员利用同位素标记实验(HO)来分析反应过程中的微量产物,发现CO中的氧原子既来源于HO,也来源于O。这是因为NbO价带的空穴将HO氧化为OH,而导带的电子将O还原为O。原位电子顺磁共振光谱也显示在聚乙烯光转化过程中确实生成了OH和O自由基。此外,原位傅里叶变换红外光谱检测到了COOH中间体。基于以上结果,研究人员提出了一种顺序光诱导C—C 断键和偶联机理(图11)。首先,废塑料中的C—C键被OH和O氧化裂解为CO,与此同 时,O被 还 原 为O、HO和HO。随 后在COOH 中间体的作用下,产生的CO通过C—C偶联反应被还原为CHCOOH。这一转化路线能实现的关键条件在于NbO的能带边缘能同时满足上述两个过程的氧化还原电势。单原子厚度的片状NbO暴露了足够多的活性位点,提升了光催化性能。虽然这项工作实现了废塑料光催化转化为有价值的化学品的概念验证,但在这项技术真正实现工业化之前仍有一些问题需要解决。首先,单原子厚度的片状NbO单批合成的产率较低,不利于大规模生产。此外,单原子厚度的NbO由于其较高的表面能而会出现团聚现象,从而减少表面暴露的活性位点,进而影响其光催化性能。然后,未改性NbO的宽带隙特征导致它只能吸收太阳光谱中占比极少的紫外光,严重限制了对太阳能的利用。最后,NbO表面较差的HOOC—CO吸附能力和较弱的CHCOOH 解吸能力意味着大部分的塑料都被矿化为CO,而没有被有效地转化为更高附加值的C化学品。
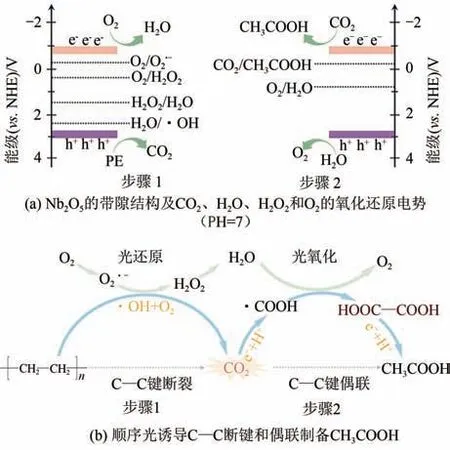
图11 Nb2O5光催化塑料制备C2化学品的反应机理
6 后聚合改性
除了将废塑料转化为高附加值的燃料和化学品,在保持聚合物骨架完整的情况下,还可以采用后聚合改性的方法赋予塑料新的功能和特性。
聚乙烯虽然结构简单,但是结晶度高,主链上缺乏活性基团,因而不具备优异的表面黏附性和韧性。极低的极性使它也几乎不能和其他极性单体共聚。加入自由基引发剂虽然能对聚乙烯链段进行后处理,但会引发链段的交联或者降解,导致加工性能和力学性能显著下降,因此对C—C 键进行后改性存在牺牲材料性能的风险,C—H 键的改性仍然是唯一合理的方案。Hartwig 等设计了一种多氟化钌卟啉催化剂,只需要加入0.02%(摩尔分数)的催化剂,在120℃下的二氯乙烷溶液中0.5h 就能实现聚乙烯C—H键的选择性氧化,引入羟基或者羰基官能团。为了研究这种多氟化钌卟啉催化剂的普适性,研究者对不同结构的聚乙烯(LDPE、HDPE、LLDPE)进行了选择性氧化。研究结果发现,尽管这些聚乙烯的结构和支化程度不同,但都能实现亚甲基的氧化。而且,通过改变催化剂的用量可以很容易地调节聚乙烯中氧化官能团的含量。此外,改性前后的聚乙烯的分子量、分子量分布、热稳定性、玻璃化转变温度和力学性能几乎没有变化,说明多氟化钌卟啉催化剂只是在聚乙烯链段上增加了含氧官能团,并没有显著破坏链结构。相较于未改性的LDPE,氧化LDPE对铝的黏结性提高了20倍。以氧化聚乙烯作为中间层黏结HDPE/HDPE或者HDPE/铝,其强度也分别增加了5 倍和10 倍。水性乳胶漆在氧化聚乙烯上的黏附强度也能显著提高。

7 结语
本文主要总结了近年来催化热解、化学解聚、催化氢解、光催化和后聚合改性等方法在废塑料资源化利用上的研究进展,指出废塑料的化学循环和化学升级再造策略为解决塑料污染问题和缓解资源危机提供了一条有效的技术途径。但是,这些新的塑料处理技术仍处于研究的早期阶段,更多地还是作为一种具有前瞻性的研究和技术储备。而且,塑料的种类繁多,并非所有类型的塑料都能用化学方法来分解。新型催化剂和催化体系层出不穷,针对不同种类的废弃塑料建立完整的升级再造技术体系仍然需要大量的基础研究工作。如果目前就将它们大规模地应用于解决塑料污染问题,那么整个工艺过程的成本和收益的平衡问题、二次产物的附加值问题以及新的二次污染物的控制问题将又是一大难题。因此从总体来看,目前已经发展比较成熟的传统催化裂解技术仍是解决较大规模废塑料污染的主要途径。但是,为了未来更好地对废弃塑料进行循环利用,半间歇或连续流动反应器的设计和优化是从实验室迈向工业化规模的关键步骤。此外,开发更加高效、环保和低成本的催化剂也是科研人员所应关注的焦点。
