水相生物油原位汽化-催化重整制氢工艺优化
2022-04-12张安东李志合王丽红王绍庆梁昌明万震
张安东,李志合,王丽红,王绍庆,梁昌明,万震
(1 山东理工大学农业工程与食品科学学院,山东 淄博 255000;2 山东理工大学山东省清洁能源工程技术研究中心,山东 淄博 255000)
我国生物质资源丰富,生物质中富含C、H、O 元素,通过热解液化技术将生物质转化为生物油,被认为是取代化石能源的有效途径之一。水相生物油是从热解油中进一步分离获得的轻质组分,其含水量高达80%左右,热值低,同时含有酸、醛、醇、酮、酚、酯、糖等几十种组分。Bridgwater认为水相生物油的主要出路是萃取提纯高价值化学品或者催化重整制氢。传统的催化重整制氢工艺中原料需先进行汽化,然后喂入反应器发生反应。然而水相生物油中的组分沸点各不相同,在汽化过程中受水沸点影响,部分高沸点组分难以快速汽化逸出,使得原料转化效率大幅降低,更严重的会发生结焦堵塞喂料管。
目前国内外关于生物油催化重整制氢的研究主要以固定床反应器为主,汽化后的生物油(模化物)通过静止的催化剂床层发生反应。优点是操作简单,易于控制,但催化剂易失活,需要卸料后重新装载催化剂。因此也有部分研究人员选择在流化床反应器上开展催化重整制氢研究,通过催化剂颗粒之间的碰撞减少积炭,从而抑制催化剂失活。但流化床相比于固定床来说也存在一些弊端,如工艺复杂、催化剂易磨损、载气流量大等。
本研究基于原位汽化策略,在一体式固定床/流化床上开展水相生物油催化重整制氢实验,重点探究原位汽化体系下水相生物油的转化效率以及流化床反应器中催化剂的抗积炭行为,为水相生物油催化重整制氢的产业化推广应用提供参考。
1 实验材料与方法
1.1 实验原料
实验所用水相生物油产自山东泰然生物工程有限公司,原料主要为松木屑和杨木屑,热解温度为500℃,在线分级冷凝获得的轻质生物油经过静置分层(本批次静置了一年以上),取上层即为水相生物油,水相生物油的理化特性和组分分布如表1和表2 所示。其中水相生物油的密度()通过简易密度计测量。pH通过雷磁数显pH计(PHS-3C)测量,测试温度为室温25℃,精度为0.01,测试前采用pH=4.00 和pH=6.86 的标准溶液对pH 计进行校准。含水率的测定是通过卡尔费休水分测定仪(瑞士万通,KF870 型)获得的,测试前需用色谱级乙醇进行稀释(100 倍),然后对测量结果进行换算获得,因为卡尔费休对含水率较大的样品测量精度较低。元素组成采用意大利Euro Vector公司生产的EA3000 元素分析仪进行分析,每次测试前需按照一定浓度梯度称取5+3个标准样品用于建立校准曲线,校准曲线的线性回归方程≥0.998则数据可用。液体样品每次取样1~3mg 放入专业锡杯中,并用封口器进行密封,测量结果三次取平均。水相生物油的组分分布通过气相色谱-质谱联用仪(GC-MS,美国Agilent 公司,6890/5973N型)进行分析。搭配DB-1701毛细管色谱柱(60m×0.25mm×0.25μm),用于分离样品中的不同组分,每次进样量为0.2μL,分 流 比60∶1, 载 气 为 高 纯 氦 气(99.999%)。进样口气化室(Inlets) 的温度为280℃,离子源(Aux)温度为250℃。色谱柱升温程序为:以5℃/min 的升温速率从40℃升至240℃,保温5min。通过NIST 17.0 质谱库识别化合物,通过峰面积百分比计算各组分的相对含量。
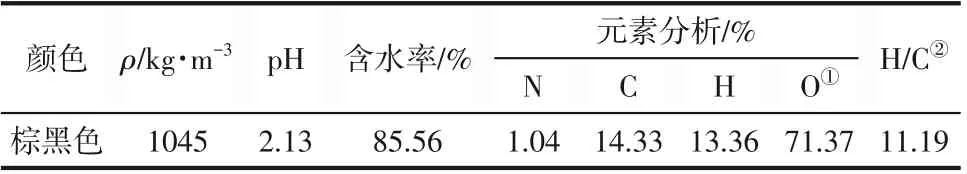
表1 水相生物油理化特性

表2 水相生物油成分分析
1.2 催化剂的制备与表征
实验所用催化剂为Ni/γ-AlO,载体为山佳硅铝新材料有限公司生产的γ-AlO微球(50~60目),Ni(NO)·6HO 作为镍源,购自国药集团化学试剂有限公司。采用过量浸渍法制备所需Ni/γ-AlO催化剂,浸渍液浓度为0.1mol/L,浸渍时间为6h(室温),过滤后干燥(105℃,12h),然后置于马弗炉中煅烧(600℃,4h),即可获得催化剂前体NiO/γ-AlO,实验前需通H将NiO 还原成高活性的镍单质(根据前期研究发现,900℃下还原30min 可以实现催化剂完全还原)。经X 射线荧光光谱分析(XRF,日本理学,ZSX-100e型)所制备催化剂中镍负载质量分数为4.3%。
反应后催化剂通过场发射扫描电子显微镜(SEM,美国FEI,sirion200型)观察其表观形貌和积炭情况,为增强材料导电性,催化剂表明进行喷金处理;通过同步热分析仪(STA,德国耐驰,449 F5)对反应后催化剂进行程序升温氧化,分析其表面积炭类型和催化剂失活原因,升温程序为40~1000℃,升温速率10℃/min,空气气氛。
1.3 原位汽化工艺设计
典型的催化重整制氢工艺往往是将原料(化石燃料、生物油、模化物等)和水预先进行汽化,然后喂入反应装置与催化剂接触并发生反应。水相生物油中含有大量的水分,可以为反应提供足够的氢源而无需额外添加水蒸气,简化了喂料过程。但由于水相生物油组分沸点的差异性,其在传统汽化器中面临梯度汽化、组分结焦等问题,因此,本研究设计将喂料管伸入到反应管恒温区,原料(水相生物油)到达喂料管末端时,利用反应器高温实现全组分的快速汽化,并在载气的携带下迅速与催化剂接触发生反应。
实验采用单一管路液体喂料,喂料管(内径=1.5×10m)伸入反应管内一段距离,利用反应管内高温进行预热并使其在离开反应管出口时瞬间汽化。这属于传热学中流体流经单一管槽内部强制对流换热的问题,针对这一问题作如下假设。假设一,水相生物油中主要成分是水,水相生物油所有热物性参数均参照水来计算;假设二,喂料管内壁温度近似等于环境温度;水相生物油温度为室温′。则喂料管出口液体温度″主要受环境温度、液体物理特性(密度、运动黏度、定压比热容c、热导率和普朗特数)、喂料速率以及反应器恒温区内喂料管长度等因素影响。
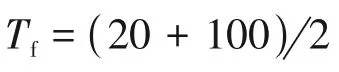
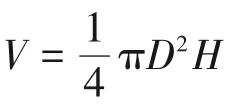
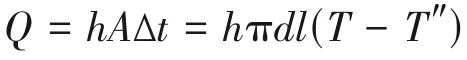
即喂料管伸入反应器恒温区长度应大于1.156mm,考虑到水相生物油组分及热物性与水之间存在一定差异,在上行式流化床[如图1(b)]上进行喂料测试,将汽化段喂料管长度设置为2mm,结果发现运行一段时间后载气流速出现剧烈波动,说明水相生物油汽化不完全,液体回流堵塞进气管。逐渐增加喂料管长度发现,当喂料管长度为5mm 时,可以实现水相生物油的完全汽化,长时间喂料载气流速依然保持稳定。因此,本研究设定喂料管汽化段长度为5mm。
1.4 水相生物油原位汽化-催化重整制氢固定床/流化床对比实验
为了探究固定床和流化床两种工艺方法对水相生物油催化重整制氢的影响,设计并加工一套适用于水相生物油原位汽化-催化重整制氢的固定床/流化床反应装置,如图1所示。该装置采用相同的喂料系统,相同的加热、保温和控温系统,相同的冷凝系统以及相同的产物分析方法。实验所用催化剂为Ni/AlO微球(60~70目),催化剂装填量均为3g[直径15mm,高(30±1)mm],实验开始之前均用10%H/N对催化剂进行还原(900℃,30min),还原结束后通入高纯氮气排净装置内的H,并将反应器温度设定为800℃。固定床实验开始之前只需将氮气流速直接设定为1000mL/min;而流化床实验开始之前需逐渐增大氮气流速至完全流化状态(冷态实验表明最佳流化速度约为1000mL/min)。待系统参数稳定后,通过蠕动泵将水相生物油喂入反应器中,LHSV 为10h,其中固定床为下行式,流化床为上行式。水相生物油离开喂料管出口时瞬间汽化,在载气的携带下通过反应管,与催化剂充分接触发生反应。反应后产物通过串联的两级冷凝器冷凝收集液相产物(0℃),同时流化床还需在反应管后增加两级带保温功能的旋风分离器,用于分离被载气携带而出的催化剂碎屑和残炭,不可冷凝气体通过气袋收集后分析成分,每次采样间隔为5min。实验结束时先停止喂料并将喂料管中残留的液体倒吸回烧杯,避免残留液体持续缓慢汽化参与反应,停止加热,并将氮气流速降至500mL/min,持续通氮气冷却至室温,取出催化剂后密封保存,用于后续表征。每次实验时间为180min,重复3次。
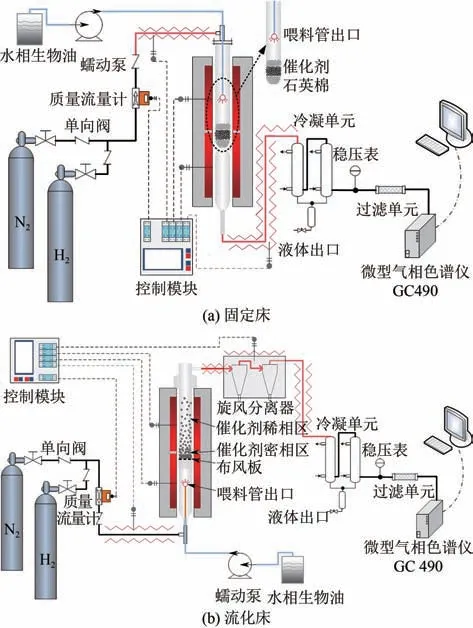
图1 水相生物油原位汽化-催化重整制氢固定床/流化床反应装置
1.5 数据处理方法
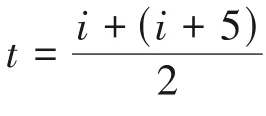
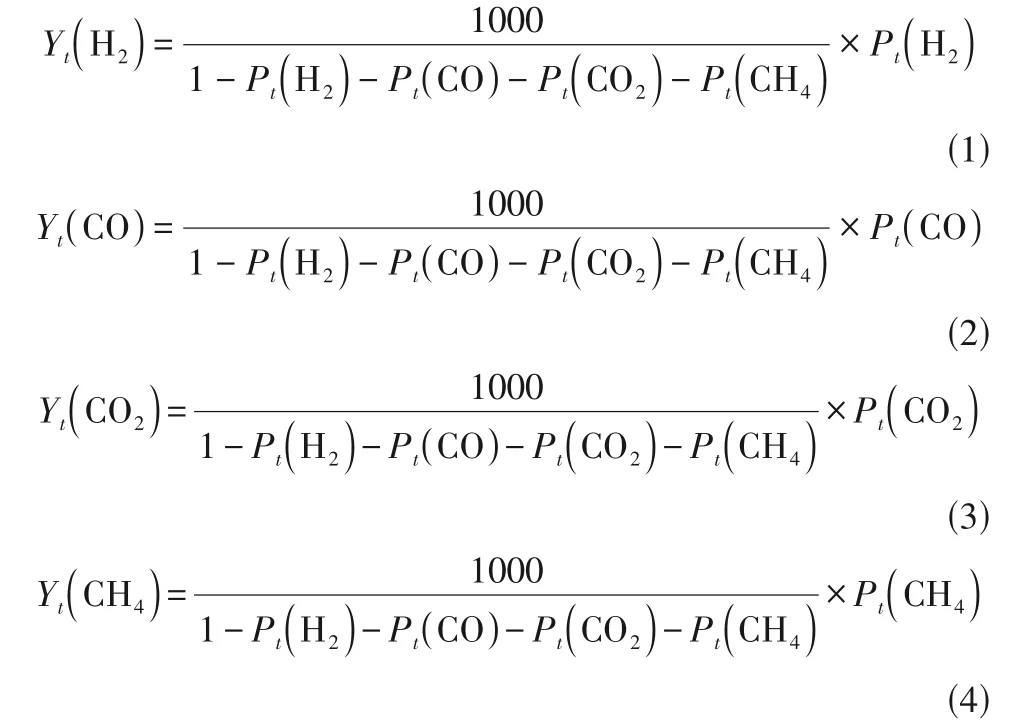
原料转化率和H选择性是评价催化重整制氢效果的关键指标。由于水相生物油为复杂混合物,因此采用碳转化率来表征原料转化效率。本实验碳转化率(X)和H选择性(S)通过式(5)和式(6)计算。
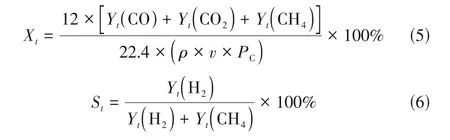
式中,为水相生物油中C元素质量分数。
2 结果与讨论
2.1 水相生物油催化重整制氢效果评价
2.1.1 实时气体产量
图2 所示分别是在基于原位汽化策略的固定床/流化床反应器上开展的水相生物油催化重整制氢实时产气量分布,从图中可以看出,基于原位汽化的两种反应器均可以实现水相生物油的高效重整制氢,产物以H和CO为主,并含有少量CO,CH产量几乎为零。在反应开始以后,两种反应器中的水相生物油均快速发生反应,产生大量H和CO逸出,其中流化床反应器中获得的H和CO产量略大于固定床反应器,实时产氢量在350mL/min左右波动,说明在流化床反应器中,水相生物油与催化剂接触得更加充分,催化重整效果更好,且由于催化剂的流化效果,使反应原料及中间产物与催化剂的接触时间(停留时间)延长,从而更好地促进了原料及中间产物的重整产氢过程。随着反应时间的进行,固定床反应器上的水相生物油催化重整过程进行到100min 左右时,其H和CO产量出现明显下降,CO 和CH的产量也逐渐上升,说明固定床反应器中的催化剂开始失活,催化重整反应活性逐渐降低。而在流化床反应器中,从反应开始后30min 左右H产量就开始逐渐降低,CO的产量也缓慢减少,说明流化床反应器中的催化重整反应效率逐渐降低,但CO 和CH的产量在180min 内并没有明显增多,说明催化剂还没有失活。待实验结束,取出催化剂后发现固定床反应器中的催化剂由反应前的3g 增加到(3.08±0.01)g,说明其在催化重整过程中产生了大量积炭附着在催化剂表面。而流化床反应器中的催化剂则只剩(1.97±0.04)g,另在旋风分离器中收集到催化剂(0.90±0.08)g,说明在流化床反应器中催化剂由于颗粒之间以及颗粒与管壁之间的碰撞磨损,造成了接近三分之一的损耗,这也是其在反应后期催化重整效率逐渐降低的主要原因。
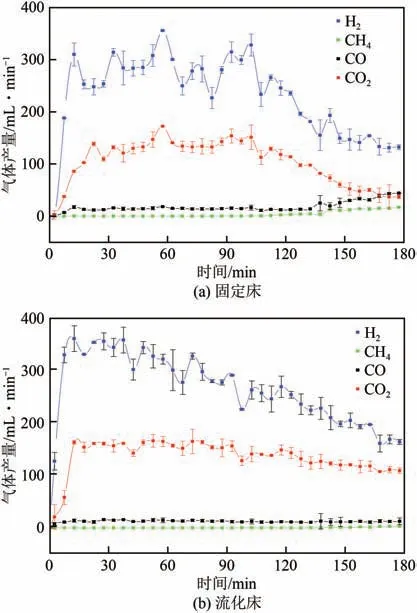
图2 固定床和流化床上水相生物油催化重整制氢实时产气量
2.1.2 碳转化率和H选择性
通过计算获得固定床/流化床反应器上水相生物油催化重整过程碳转化率和H选择性变化规律如图3所示。从图中可以看出,反应开始后水相生物油的碳转化率和H选择性均迅速升高,然后基本保持稳定。其中流化床反应器内的水相生物油碳转化率(95%左右波动)相比固定床(80%左右波动)要略高一些,说明流化床中催化剂的流化效果对水相生物油各组分的催化重整产氢过程具有明显的促进作用,增大了催化剂与反应原料的接触效率和停留时间,提高了水相生物油催化重整的转化效率。随着反应时间的延长,流化床反应器中水相生物油的碳转化率缓慢降低,根据前文分析结论,这主要是由于流化床内催化剂颗粒磨损,导致部分催化剂被携带出反应器,整体催化效果下降引起的水相生物油转化效率降低。而固定床反应器中水相生物油的碳转化率在100min 左右的时候出现一次较大幅度下降,这主要是由于催化剂的失活引起的。两种反应器内水相生物油催化重整制氢过程的H选择性从反应一开始便维持在100%,说明催化剂的催化效果和选择性非常好。当固定床中的催化剂开始逐渐失活时,其H选择性开始逐渐下降。而流化床中的H选择性始终保持在100%,说明流化床中的催化剂并没有失活,在反应最后的30min内出现的小幅降低最有可能是因为此时的喂料速率超出了流化床内剩余催化剂的处理能力,少量CH等小分子烃类来不及转化便被携带出反应管,也不排除此时流化床内催化剂出现了轻微的失活,需要做进一步的表征确定。
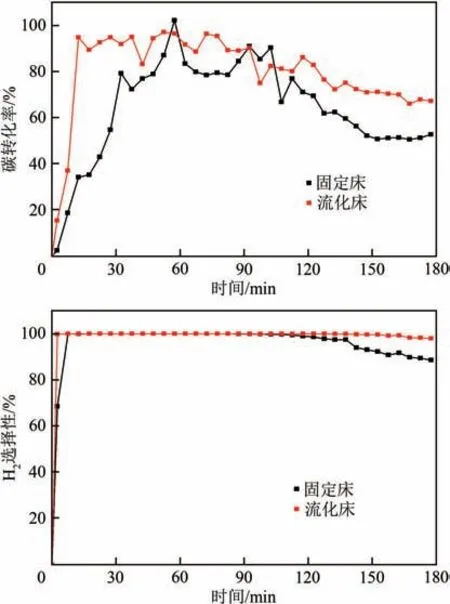
图3 两种反应器上水相生物油催化重整制氢碳转化率和H2选择性
2.2 液相产物的表征
对固定床/流化床反应器上水相生物油催化重整制氢液相产物收集后通过GC-MS 进行表征,与水相生物油组分分布进行对比如图4所示。从图中可以看出,基于原位汽化策略的催化重整转化效率非常高,水相生物油中的绝大多数组分均被完全转化,说明所采用的原位汽化策略可有效促进水相生物油全组分的汽化和催化重整转化过程。两种反应器中水相生物油经过180min 的催化重整转化过程后,均有少量苯酚残留,说明在本实验条件下苯酚的转化效果相比其他组分较弱一些,这可能是由苯环的稳定结构决定的。其中,固定床反应体系反应后收集的液相产物中苯酚含量明显高于流化床,且除苯酚以外还有少量乙酸等组分残留,未完全转化,结合前文产气量分布和碳转化率结果分析,说明在反应前期活性较高的阶段,固定床反应器中原料转化效率弱于流化床反应器,可能是由于原料在催化剂床层停留时间较短,部分原料来不及完全转化;而在反应后期,固定床体系中催化剂出现明显失活,进一步降低组分转化效率。相对而言,流化床反应体系液相产物中仅检测到很少量的苯酚残留,说明流化床中各组分转化效率非常高,这主要与流化床中较长的停留时间、较高的催化剂接触效率和抗失活特性有关。此外,在两种反应体系反应后的液相产物中均发现了一定量丙酮的生成,这主要是由羧酸(乙酸)酮基化反应产生的,而流化床反应体系液相产物中丙酮含量远低于固定床反应体系,这是因为流化床中催化剂处于流化状态,催化反应区间远大于固定床反应体系,羧酸酮基化产生的丙酮在催化剂表面进一步断键重整,使产氢量和碳转化率进一步提高。在固定床反应体系液相产物中还检测到少量2-丁酮,这同样是由酮基化反应产生的,水相生物油中的丙酸是其主要来源,但在流化床反应体系的液相产物中无法检出,说明少量的2-丁酮在本实验所述条件下可以完全转化,不会随载气逸出。
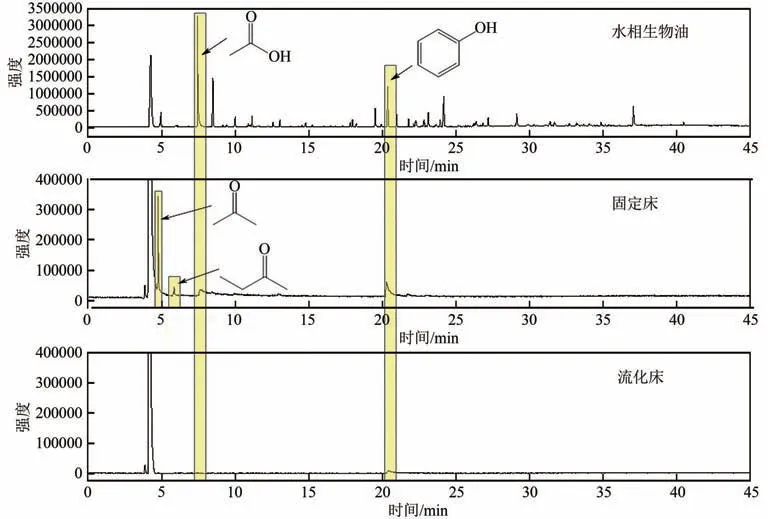
图4 液相产物色谱图
2.3 催化剂的表征
2.3.1 SEM
取固定床/流化床反应器中反应前后催化剂,通过SEM对其表观形貌进行表征,如图5所示。从图中可以看出,反应前催化剂表面均匀分散的镍颗粒发生了一定程度的团聚,这主要是由还原过程的高温导致的。经过催化重整制氢反应后,固定床反应体系中的催化剂表面被大量纤维状积炭包裹,这在之前的传统固定床催化重整实验中也同样被发现,而在原位汽化-催化重整制氢体系中,催化剂失活的时间被大幅延后,说明水相生物油的高效汽化过程可以降低积炭失活速率,但并不能阻止积炭的生成。而在流化床反应体系中的催化剂表面并没有发现明显的纤维状积炭,这也说明流化床反应体系中的催化剂在重整过程中始终保持较高活性,没有被积炭覆盖。一些学者认为流化床通过颗粒碰撞可以避免催化剂表面积炭的沉积,但本文认为流化床中催化剂表面没有纤维状积炭生成的主要原因不是颗粒碰撞,而是活性位点的转移。因为纤维状积炭是以化学键的形式在催化剂表面生长的,而非简单的物理覆盖,这通过对反应后催化剂进行简单的碰撞试验即可验证。在流化床反应体系中,催化剂表面活性位点与反应原料接触触发化学键断裂后迅速转移到下一个位置继续参与反应。在整个流化床反应区间内,流化密相区分布的主要是反应原料(即水相生物油各组分),而在流化稀相区分布的则主要是活性中间产物(高活性的游离基团)、水蒸气和终端气体产物(H、CO、CO等),这些活性自由基、水蒸气和H等终端产物会与催化剂表面形成的积炭前体发生反应,进而抑制纤维状积炭的生长,避免了在重整过程中因积炭包覆导致的催化剂失活。同时,从流化床反应体系中催化剂表面还可以看出,催化剂表面结构遭到了一定程度的破坏,这主要是由于流化床中颗粒碰撞磨损造成的,这也是导致其在催化重整反应后期产氢率和碳转化率降低的主要原因,因此开发高强度、耐磨损的重整催化剂将是后期研究的重点。

图5 两种反应器上反应前后催化剂SEM图(放大40000倍)
2.3.2 TG-DTG-DS
图6 所示为固定床/流化床反应器中反应后催化剂TG-DTG-DSC 图。从图中可以看出,流化床反应器上水相生物油催化重整制氢反应后催化剂在程序升温过程中只有一个明显的失重峰,即在77℃左右的失水峰。而固定床反应器上反应后催化剂在程序升温过程中除失水峰以外,还出现了三个失重峰,分别分布在354℃、538℃和651℃左右,其失重率分别为0.38%、6.63%和0.75%,分别对应单质碳(或吸附在表面的焦炭)、纤维状积炭(聚合碳)和碳化镍。从积炭氧化顺序可以看出,碳化镍的性质最为稳定,它是镍单质被积炭氧化生成的。而在催化剂表面聚合形成的纤维状积炭将镍颗粒包裹,使其无法与周围具有还原特性的活性自由基接触再生,是导致催化剂失活的主要原因。

图6 空气氛围下反应后催化剂TG-DTG-DSC图
3 结论
基于原位汽化策略,设计加工一套固定床/流化床催化重整一体式反应装置,对比分析水相生物油在两种反应器上催化重整制氢效果,重点探究流化床反应器中催化剂的抗积炭行为以及原位汽化体系下水相生物油的催化转化效率问题,主要结论如下。
(1)流化床反应器内水相生物油转化效率(95%左右)明显高于固定床(80%左右),且流化床气体产物中H产量略高于固定床,两种反应器中H选择性可达100%并能保持较长时间稳定。
(2)在重整反应后期,固定床反应体系中催化剂被积炭覆盖逐渐失活,而流化床反应体系中催化剂始终保持较高活性,未发现积炭。
(3)流化床反应体系中水相生物油各组分几乎完全转化,而固定床反应体系中仍有少量乙酸和苯酚残留,并伴随产生了少量酮类物质(丙酮等)。
对于稀疏解混模型,其包含了一个完备的端元光谱库,该光谱库通常是通过实验条件或者野外采集等手段获取,再将采集到的大量纯净地物光谱进行组合,里面囊括了每类端元所有可能的光谱[18]。相对于成千上万条的端元光谱库,而每个混合像元通常只是由3~5个端元构成,那么丰度是稀疏的,于是这就成了一个稀疏问题[15]。为此,结合稀疏表示理论,利用完备的端元光谱库构造用于稀疏分解的过完备字典,通过对丰度进行稀疏约束,将解混问题转化为稀疏回归问题进行求解。
(4)基于原位汽化策略的水相生物油催化重整制氢过程简化了生产工艺,降低生产成本的同时大幅提高了生物油的重整转化效率,结合流化床中催化剂流化效果,可实现水相生物油重整制氢的连续化运行,有助于加快生物质热解液化-生物油催化重整制氢产业链的发展进程。
——喂料管横截面面积,m
c,c′,c″——原料的定压比热容以及20℃和100℃下水的定压比热容,kJ/(kg·K)
——反应管内径,m
——喂料管内径,m
——催化剂装填高度,m
——单一管槽内对流传热表面传热系数,W/(m·K)
LHSV ——液时空速,h
——反应管恒温区内喂料管长度,m
——单位时间内流经喂料管内单位横截面积原料/水的质量,g
——单一管槽内对流换热努塞尔数
——水相生物油中C元素质量分数,%
——原料/水的普朗特数
P——时刻气体产物中某组分的体积分数,%
——液体升高一定温度吸收的热量,W
——液体流动的雷诺数
S——时刻气体产物中H的选择性,%″——环境温度、室温、喂料管出口液体温度和换热计算定性温度,℃
——催化剂体积,m
——原料/水的喂料速率,m/s
′ ——喂料管内单位横截面积原料/水的喂料速率,m/s
X——时刻原料的碳转化率,%
Y——时刻气体产物中某组分的产量,mL/min
——原料/水的热导率,W/(m·K)
——原料/水的运动黏度,m·s
——原料/水的密度,kg/m
——取样中间点的时间,min
