CO2电催化还原产合成气研究进展
2022-04-12华亚妮冯少广党欣悦郝文斌张保文高展
华亚妮,冯少广,党欣悦,郝文斌,张保文,高展
(1 西安交通大学化学工程与技术学院,陕西 西安 710049;2 佛燃能源集团股份有限公司,广东 佛山 528000)
2020年,我国提出的“碳达峰”“碳中和”目标对CO减排提出了更高的要求。电催化CO还原反应(COreduction reaction,CORR)是实现CO资源化利用,使其转化为具有高附加值化学产品的一种有效方式,对减少温室气体排放具有重要意义,吸引了全球科学家们的重点关注。CO电催化还原反应在室温常压下进行,所需的电能可直接从太阳能、风能、地热能、潮汐能等可再生能源中获取,是一种将绿色能源转化为化学能的高效储能方式。CORR 是一个涉及多质子耦合多电子转移的过程,通常包括2e、4e、6e、8e、10e、12e、14e等反应路径,还原产物众多,有C产物(CO、CH、HCOOH、CHOH)、C产物(CHCH、CHOH、CH、CHCOOH、CHCHO)以及C产物(CHCHO、CHOH)等。表1 总结了在标准大气压和25℃下部分CORR 产物及其在中性水溶液中相对标准氢电极(standard hydrogen electrode,SHE)的平衡电势。

表1 电化学还原CO2半反应的电极电位(标准试验条件)[10]
1 CO2 电催化还原产合成气面临的挑战
CO电催化还原时,当以水溶液为电解质,由于阴极电解水发生析氢反应(hydrogen evolution reaction,HER),而HER 具有与CO电催化还原反应相当的热力学电势,因此在阴极CORR 与HER属于竞争关系。当调控阴极CORR和HER反应时,则可以选择性地生成CO和H的混合气,即合成气产物,它是石油化工行业重要的合成原料,可用于费托合成或生产甲醇等。传统制备合成气的方法包括煤气化和天然气重整等,需在高温、高压等极端条件下进行,消耗不可再生能源,与绿色化学的理念不符。利用CO和HO 作为原料,在水溶液中电催化还原CO,是可持续制备合成气的理想方法,无需抑制析氢反应,也没有液态产物分离困难等问题,成为当下的研究热点。
尽管CO电催化还原制备合成气在能源和环境方面具有独特的优势,但由于CO分子是一种热力学稳定的线性分子,键长短、键能高、化学性质相对惰性,因此采用电化学方式还原CO产合成气时,存在CO活化能高、反应过电势高、选择性差、催化活性低等问题。另外,由于不同的化工过程,所需的CO 和H组成比例不同,所以在CO电催化还原反应过程中调控合成气组成比例,对下游合成气的工业化应用极为重要。但目前仍然很难保证在高电流密度的同时实现宽范围调控合成气组成比例。因此,开发经济、稳定、高效的电催化剂,通过催化剂结构设计,降低反应过电势,提高催化活性,实现宽范围精准调控合成气组成比例,是推动电催化CO还原产合成气技术应用的关键所在。
另外,水溶液中电还原CO产合成气反应发生在气、液、固三相界面处,涉及CORR 及HER 两个反应,反应过程包含:①电解液中反应物CO向电极表面扩散;②CO和HO 被催化剂活性中心吸附;③电子和质子通过催化剂传递到催化剂表面的同时CO发生加氢加电子反应,产生COOH、CO、HO、H 等中间产物;④中间产物进一步转化为CO、H等最终产物;⑤产物从催化剂表面解析等(如图1所示)。因此,催化剂表面电子传递、加氢反应过程以及气、液相传质等都是CO电催化还原产合成气反应的关键。根据Sabatier 原理,中间态物质与催化剂表面结合(化学吸附)既不太强也不太弱时,才会具有最佳的催化活性。因此,通过调节催化剂固有电子结构,获得其表面对COOH、CO及H等中间产物最佳的化学吸附特性,调控CO 和H组成比例,可实现高效的CO电化学还原产合成气反应催化活性。由此可见,CO电催化还原制合成气反应的催化剂应当满足以下要求:①对关键中间体COOH 具有适宜的吸附强度;②适宜的CO 吸附能;③CO 还原能力较弱;④原料丰富,价格低廉;⑤效率高,能够同时生成CO和H。
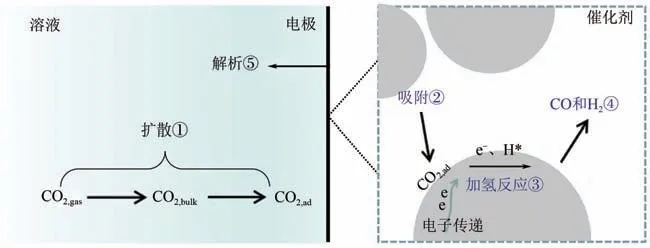
图1 电极表面电催化反应过程
基于此,本文综述了电催化还原CO产合成气反应的催化剂研究进展以及用于电催化反应的电化学池特征。进一步地,总结了提高电催化还原CO产合成气效率的策略,包括阳极反应耦合增强反应效率、设计双活性位点电催化剂协同催化CORR、催化剂多级形貌调控增强反应传质过程等,为电催化还原CO产合成气技术工业化应用提供理论支持。
2 CO2电催化还原产合成气催化剂
根据以上分析,由于标准条件下生成H和CO的电极电势相似,在CO-HO 体系中,CO 和H都是电解产物。然而,催化剂的性质(如元素组成、结构、表面形貌、颗粒大小)和操作条件(如电压、温度、压力、电解质的pH 等)对调节合成气比例、提高合成气生成效率起着重要的作用。几十年来,开发高效、廉价、适宜的CO电催化还原产合成气反应催化剂已然成为研究热点。通过调节催化剂的大小、结构和组成,能够提高催化剂的催化性能。在本节中,将讨论5种不同类型的用于水溶液中CO电催化还原产合成气的催化剂特征。
2.1 金属催化剂
2.1.1 贵金属催化剂
电催化CO还原产CO的催化剂中,由于Au和Ag 等贵金属与CO 的结合能适中,不能进一步将CO还原为其他碳氢化合物,所以表现出较好的CO选择性和催化活性。Hansen等构建了一个基于密度泛函理论计算的模型,通过催化剂对反应中间体CO和COOH的吸附能来描述CO电还原成CO的催化活性趋势。由图2(a)可以看出,Au 和Ag 具有较强的COOH 吸附能,同时具有较弱的CO 吸附能,从而表现出较强的CO电催化活性。水溶液中Au 催化CO生成合成气的选择性、活性以及合成气组成比例等都与Au 纳米颗粒的尺寸大小密切相关。Zhu 等研究了粒径分别为4nm、6nm、8nm、10nm 的Au 纳米颗粒电催化还原CO产CO 性能,结果表明,粒径为8nm 时,Au 催化CO还原生成CO的选择性最好[图2(b)],且当粒径分别为4nm、6nm、8nm、10nm 时,CO 和H比例分别为6∶4、7∶3、9∶1和3∶1。密度泛函理论计算结果表明,边缘位点能够催化生成CO,而拐角处的位点则是HER 反应的活性位点。边缘与拐角位点的比例及活性位点数量均与Au粒径大小息息相关。当Au纳米颗粒粒径大小不同时,其表面能和配位数不同,因此生成CO 和H的比例也不同,且当Au 纳米颗粒粒径越小、活性位点数越多时[图2(c)],催化活性越强。

图2 金纳米颗粒催化活性
对于Au电极来说,其电催化CO还原生成合成气的选择性和活性,除与粒径大小相关外,还受晶界(grain boundaries,GB)参数的影响。Feng 等采用气相沉积法制备了Au/CNTs催化剂,其表面晶界密度受煅烧温度影响。研究结果表明,Au/CNTs催化剂表面晶界密度与其催化CO还原产CO 选择性呈线性相关,因此可以通过改变催化剂表面晶界密度调节合成气产物组成比例。另外,合成气组成比例(CO∶H)也可以通过改变Au纳米线长度来调节。例如,当Au纳米线长度分别为15nm、100nm、500nm,在电解电位(.RHE)为-0.4V 时,相应的CO∶H比例分别为1∶3、3∶1和9∶1。相比于Au 电极,Ag 电极因其具有较强的CO还原活性,价格较Au电极便宜,研究也较为广泛。Liu等研究了三角形貌的Ag纳米颗粒,其电催化CO还原,表现出高的CO选择性和催化活性,CO的法拉第效率高达96.8%,原因在于三角形貌的Ag 纳米颗粒具有更多的Ag(100)晶面,并且具有适宜的边缘和转角位点比例,合成气的组成比例可以通过Ag 纳米颗粒的形貌来调节(图3)。

图3 银纳米颗粒制备过程及催化机理[30]
近年来,关于CO电还原催化剂结构设计已经从抑制HER 反应到调控CORR 和HER 反应活性产合成气,调节合成气组成比例。Sheng 等利用商用Pd/C纳米颗粒在水溶液中电还原CO生成组成比例可调的合成气,当电解电位(.RHE)在-0.5~-0.6V时,得到CO/H比例在0.5~1之间,满足工业上费托合成的需求。并且他们结合原位同步辐射和原位XRD 等,证实了Pd 在反应过程中与H 结合转化成PdH的过程,PdH的形成能够极大降低催化剂对中间产物CO 和H 的结合能,从而调节CO 和H的生成。Liu 等合成了过渡金属氮化物负载的钯纳米颗粒(Pd/TMN)用于电催化CO还原产合成气,取得优于商业Pd/C 的催化活性。通过原位XRD 及理论计算等证实,过渡金属氮化物作为载体材料能够有利于Pd 到PdH 的相转变,PdH 的形成能够降低CO 吸附能,避免Pd 表面CO 中毒。根据研究结果,5%的Pd/TMN 催化活性相当于10%的商用Pd/C,以过渡金属氮化物为载体,显著增强了Pd的催化活性和选择性。
2.1.2 非贵金属催化剂
由于贵金属资源稀缺、价格贵,不利于大规模应用。因此,近年来,关于非贵金属催化剂电催化CO还原产合成气的研究越来越多。Qin 等采用电化学沉积法在泡沫铜表面合成了Zn 纳米催化剂[图4(a)],用于电还原CO产合成气,在电解电位(RHE)为-0.7~-1.3V、温度40℃、KHCO浓度0.1mol/L 时,生成CO/H的比例为0.5~1.2,电流密度11mA/cm[电解电位(.RHE)为0.9V]。根据研究结果,Zn(101)晶面表现出催化CO还原产CO 活性,而Zn(002)晶面则更容易发生析氢反应,因此,可通过调节Zn(101)与Zn(002)晶面比例来调节合成气组成比例。Jeon等研究结果表明Zn纳米颗粒催化活性和选择性受催化剂粒径大小影响较为显著,当Zn 纳米颗粒粒径为3~5nm 时,Zn 催化剂表现出高催化活性,CO 产物法拉第效率高达70%以上,小于3nm 时主要为析氢反应,而大于5nm时,其性能类似于块体锌电极特性[图4(b)]。计算结果表明,纳米颗粒粒径越小,其表面氢的覆盖度越高,越易生成氢气,通过调控Zn 纳米颗粒粒径,即可实现合成气组成比例的宽范围调节。
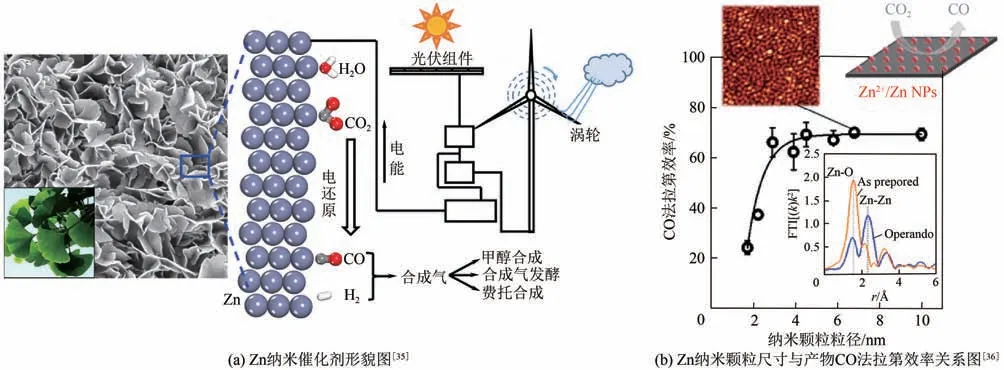
图4 锌纳米颗粒电催化还原二氧化碳
虽然非贵金属催化剂在电催化还原CO产合成气领域有了一定的应用,但目前高效的催化剂主要还是贵金属催化剂,非贵金属催化剂的催化活性依然偏低,合成气法拉第效率低,反应过电位高,电流密度低,且难以宽范围调控合成气组成比例。因此,仍需开发新型非贵金属催化剂,提高催化活性,同时实现宽范围调控合成气组成比例。
2.1.3 合金催化剂
研究表明,多组分催化剂表现出与单组分不同的催化性能。由于不同组分之间的相互作用,物理和化学性质发生了改变,从而表现出不同的催化性能。合金可以调节催化剂电子结构及形貌特征,调节催化剂选择性和催化活性,然而精确控制合金组成比例依然面临很大挑战。Fan 等通过在较大的负电流中进行电化学沉积,合成了SnPb合金催化剂。该合金催化剂具有多孔蜂窝状主体结构和树枝状次级结构,CO电化学还原起始电压比树枝状的Pb低80mV,在-1.16~-1.56V的电压区间内生成甲酸法拉第效率接近100%。通过DFT 计算表明合金材料表面HCOO中间产物的吸附能较低,所以它的起始电压较低。
通过合金催化剂还原CO和HO生成合成气的研究报道相对较少。Watanabe 等在1991 年通过电镀技术首次制备了双金属的铜合金(CuNi、CuSn、CuPb、CuZn、CuCd 及CuAg)催化剂,用于CO电还原产合成气。研究结果发现,通过将低氢过电位的金属位点引入高氢过电位的金属表面(或反之),使反应物CO和H被吸附在相邻的金属位点上,从而降低反应过电位,调节CO和H组成比例。另外,过渡金属(Fe、Co、Ni 等)与Au 的合金催化剂同样能够催化CO还原产合成气,并且调节CO和H比例。Ross等合成了Fe基、Co基及Ni 基修饰的Au 电极电催化CO还原产合成气,CO和H比例可以通过改变过渡金属与Au的组成比例来调节。DFT 计算结果表明,Au 表面更有利于吸附COOH 中间产物,而过渡金属元素3d 轨道更有利于发生HER 反应。该合金催化剂既能在更宽的电势范围调节合成气组成比例,又能显著增大反应的电流密度。
除了上述的二元合金外,三元合金也表现出较强的CO电催化能力,并且能够调节CO 和H组成比例。He 等制备了硫硒镉(CdSSe)合金纳米棒催化剂,电流密度能够在较宽的电势范围内都保持在10mA/cm以上。当CdSSe合金纳米棒催化剂中不含Se时,CdS纳米棒电催化CO还原,在电位(.RHE)为-1.2V时CO产物的法拉第效率最高达81%,电流密度为27.1mA/cm。随着CdSSe合金纳米棒中Se 含量逐渐增加,H的法拉第效率逐渐增大。Xu等合成了MoSeS合金催化剂,同步辐射结果表明,缩短的S—Mo 键和拉长的Mo—Se 键都有利于调节Mo 原子的d 带电子结构。能量计算表明,Mo 原子周围的偏心电荷不仅有利于COOH中间体的稳定,而且有利于CO 脱附。MoSeS 合金单分子层在电解电位(.RHE)为-1.15V时CO产率最高,法拉第效率达到45.2%,远高于MoS单分子层(16.6%)和MoSe单分子层(30.5%)。研究结果揭示了三元过渡金属TMDs合金中部分离域的电荷在原子水平上增强了催化剂电催化性能,为CO电还原催化剂开辟了新的研究领域。
2.2 金属配合物催化剂
近年来,金属配合物催化剂被认为是一类最具竞争力的电化学还原CO产合成气反应催化剂,原因有:①金属配合物催化剂(分子催化剂)对于一个既定反应过程表现出极好的活性、稳定性以及选择性;②分子催化剂具有清晰的结构,能够人为地设计和优化,达到更好的性能;③将分子催化剂负载在各种载体上可以用于制备单分散异相催化剂。非贵金属配合物催化剂表现出优异的电催化CO和HO 还原制合成气的能力,早在1980 年,Fisher等制备Ni、Co配合物催化剂用来在水溶液中电化学还原CO产合成气,采用Co 配合物作催化剂时,CO 和H总的法拉第效率可以达到94%。Wang 等利用Ni(Ⅱ)三齿配合物均相催化剂[图5(a)、(b)]电还原CO和HO,当电解液(DMF/HO)中含有5%的HO时,生成H的量可以忽略,主要产物为CO,当调节电解液中HO 的含量以及改变电解电位时,可以调控产物中CO 和H的组成比例,转化频数达1.9×10,催化剂可持续稳定反应一天。催化反应机理如图5(c)所示。

图5 镍配合物电催化还原二氧化碳[46]
贵金属配合物催化剂同样具有较高的电催化CO还原产合成气催化活性。Wang等巧妙设计了双功能的Ru 配合物催化剂[图6(a)],阴极室中电催化还原CO和HO生成合成气产物,同时阳极室中苯甲醇被氧化为苯甲醛[图6(b)]。如图6(c)所示,Ru(0)配合物作为反应中间物,不仅能与CO反应生成CO,还能与水反应生成H。CO 和H的组成比例可以通过调节电解液pH 以及电解电位来调控。

图6 钌配合物催化剂电化学还原CO2产合成气[47]
2.3 金属氧化物和硫化物催化剂
电子从电子给体转移至CO被认为是CO活化的关键步骤,因此通过调节催化剂表面电子结构来改善其催化活性是一种重要途径。近年来,通过在非贵金属催化剂中构建阴离子空位的方式来提高催化活性和调节合成气组成比例受到了广泛的关注。Qin等制备了硫化镉修饰的碳纳米管复合材料,其电催化CO还原生成CO 法拉第效率高达95%,通过电子顺磁共振(EPR)及原位差分电化学质谱法等证实了在CORR 反应过程中,催化剂CdS-CNT 表面原位生成了硫空位。随着硫空位数量增加,其电催化CORR 催化活性增强,电荷转移阻抗显著下降。结合原位红外光谱及DFT 理论计算结果可知,硫空位能够改变催化剂表面电子密度,降低中间产物COOH到CO转变的能垒。因此通过改变硫空位的数量可调节CO和H的比例。同样地,氧化物中的氧空位也能够调节CO∶H比例。Geng等在富电子的催化剂表面通过引入氧空位来有效促进CO的活化。采用富含氧空位的ZnO纳米片电催化CO还原产CO,电流密度高达-16.1mA/cm,CO 法拉第效率83%。理论分析表明,氧空位的引入增加了ZnO 的电荷密度,增强了CO的结合能,从而使得CO活化增强。
在以硫化物和氧化物作为催化剂电还原CO方面已有大量报道。Asadi 等证实二硫化钼(MoS)具有较强的CO催化活性,MoS边缘Mo原子因其金属特性以及较高的d带电子密度,使得催化剂具有较高的催化活性。通过调节电位(.RHE)-0.2~-0.8V,使得CO 和H的比例在1~99 之间得到有效调节。Rahman Daiyan 等利用火焰喷涂热解法制备了含有晶体缺陷的ZnO纳米材料,在连续流反应器中电催化还原CO产合成气,当槽压为2.6V 时,合成气比值为1,电流密度可达40mA/cm。
2.4 金属单原子催化剂
近年来,氮掺杂碳负载过渡金属催化剂(MN-C,M=Fe、Co、Ni、Cu、Zn、Mn等)因价格低廉,具有CO电催化还原活性,成为最具潜力的贵金属催化剂替代品之一。根据负载型金属纳米催化剂的尺寸效应,金属尺寸的减小不仅能够增加金属位点的不饱和配位环境,增强金属本征活性,而且又能增强金属与载体及吸附物之间的相互作用,提高催化活性。当金属尺寸缩小至极限即单原子时,其催化活性呈指数增长,因此,原子级分散的M-N-C催化剂成为近年来CO电催化剂的研究热点之一。原子级分散的M-N-C 催化剂,作为一种单原子催化剂(single atom catalysts,SACs),相比于块体及纳米催化剂,具有原子级高度分散的活性位点、配位不饱和度高、均一的配位环境等特点,使其在电催化CO还原反应中呈现出高原子利用率、高催化活性及选择性等优势,表现在:①SACs表面均一的特性使得其对CO和H产物具有优异的选择性;②催化剂表面原子利用率可达100%;③在合成气形成过程中有利于检测活性位点及反应路径。
近几年来,研究者们在过渡金属M-N-C 单原子催化剂电催化CO还原产合成气方面取得了较大的突破。通常过渡金属纳米颗粒具有较好的HER活性,因其资源丰富,廉价易得,成为贵金属Pt催化剂最具潜力的替代品之一。研究发现,当过渡金属元素(Fe、Co、Ni 等)以单原子形式均匀分散在载体上时,其具有极强的电催化CO和HO生成合成气的活性。Yang 等通过热解法制备了Ni 单原子催化剂,通过高角度环形暗场扫描透射电子显微镜(HAAD-STEM)表征了Ni原子尺寸只有0.2nm,Ni 单原子价态在0~2 之间。相比于氮掺杂石墨烯负载的金属镍纳米颗粒,氮掺杂石墨烯负载的Ni单原子催化剂电催化CO还原过电位下降了200mV,当电解电位(.RHE)为-0.95V 时,CO和H的比例为9∶1,远高于Ni 纳米颗粒。Ni 单原子催化剂稳定性长达100h。Song 等利用Co-CN单原子催化剂电还原CO,当电解电位(.RHE)在-0.7~-1.0V 时,合成气法拉第效率可达100%,CO/H的比例为0.5。
另外,金属单原子价态也会影响其催化CO还原产合成气性能及合成气组成比例。Gu 等报道了单原子分散的Fe-N-C 催化活性远高于单原子分散的Fe-N-C,研究结果表明,三价态的Fe 在N掺杂碳载体中能够与吡咯态氮配位,在催化反应中一直保持Fe形式。当CO 部分电流密度为1.0mA/cm时,Fe-N-C 需要的电位(.RHE)为-0.525V,而Fe-N-C需要的电位(.RHE)仅为-0.285V。另外,当电解电位(. RHE)从-0.2V下降到-0.45V 时,Fe-N-C 电极能够调节CO∶H的比例为(2∶1)~(18∶1),而Fe-N-C 电极调节的CO 和H比例仅为2~4。相比于Fe,Fe具有更强的CO吸附能力,而对CO 的吸附较弱,因此有利于CO生成。
由于单原子负载型催化剂中,金属单原子与载体间存在强相互作用(strong metal-support interaction,SMSI),可使电子在载体和金属之间发生传递,从而影响催化剂表面化学吸附性能,另外,通过调控载体的多孔结构,有利于改善物质传输性能。因此,作为单原子催化剂载体须具备以下几个方面的功能:①具有能适合反应过程的形状和大小;②有足够的机械强度和抗拉强度,能够经受反应过程中的机械冲击;③有足够大的比表面积,以均匀负载足够多的活性中心;④合适的孔结构,以利于气、液、固等多种形态的反应物和产物的传递过程;⑤稳定性好,以抵抗活性组分、反应物及产物等的化学侵蚀。
2.5 非金属催化剂
快速发展的碳材料因其自然资源丰富、稳定性好以及环境友好,被认为是最具潜力的产合成气非金属电催化剂。另外,碳材料不仅具有较大的比表面积、超高的电导率,并且具有多孔结构,孔隙率高,从而有利于CO的吸附以及电解液的快速渗透。但是天然的碳材料具有较低的活化CO和HO分子的能力,而热解的碳材料通常对于共还原CO和HO活性较低。然而,当掺杂了其他杂原子(N、S、B、O 等)时,会调节碳材料内部电子结构,从而表现出优异的产合成气催化活性。Kumar 等首次报道了非金属碳材料作为电催化剂同时还原CO和HO,通过静电纺丝技术利用聚丙烯腈(PAN)制备了碳纳米纤维(CNFs),采用CNFs 作为催化剂电催化CO,当电解电位(.SHE)为-0.573~-1.14V 时,产生合成气产物,其中,吡啶氮和三维石墨烯结构在催化反应过程中起到了关键作用。Ji等采用等离子体技术处理的氮掺杂碳纳米管阵列,作为电催化剂,通过电化学CO-HO 还原制备CO/H比例可控的合成气产物。在不同的等离子体处理条件下,CO/H比例范围可达0.55~3.03,适用于下游化工生产的原料气标准。通过优化等离子体处理条件,CO 的法拉第效率最高可达75%,并且能够维持稳定性长达10h。根据研究结果可知,碳纳米管中掺杂的吡啶氮有利于使CO转化为CO,而吡咯氮和sp2平面外的碳则有利于H的产生。因此,利用等离子体处理的方法能够有效调节催化剂中各活性组分的比例,从而调控CO还原反应和HER 的速率,最终实现CO/H比例可控的合成气产物。
通常认为在杂原子掺杂的碳材料中,杂原子为催化活性位点,但近期的研究报道指出,被杂原子活化的碳原子是电催化CO还原产合成气的活性中心位。Xie 等研究报道了在以F 掺杂的碳电极电催化还原CO时,F 元素的掺杂能够活化与其邻近的碳原子,一方面有利于增强中间产物COOH 在电极表面的稳定性,另一方面能够抑制HER过程,从而有利于CO到CO 的转变过程。当电解电位(.RHE)在-0.5~-1.0V 时,纯碳材料电还原CO时没有活性,而对于F 元素掺杂的碳材料电还原CO时,可以调节CO 和H产物的比例,得到合成气组成比例在(3∶14)~(9∶1)之间。
3 CO2 电还原产合成气的电化学反应池
3.1 H型电解池
CO电还原反应通常在三电极体系中进行,即采用H 型电解池(图7),中间采用离子交换膜将阴阳极室隔开以利于CO还原产物的收集检测,同时避免产物的再氧化。阳极电解水生成氧气,即发生氧的生成反应(oxygen evolution reaction,OER),阴极室CO接受电子和质子发生还原反应,同时,阴极电解水生成氢气。但由于水溶液中CO溶解度低(0.033mol/L),在传统的H型电解池中传质受限,使得CO电还原产合成气反应电流密度低,一般不超过100mA/cm,使该技术在实际应用中受限。

图7 典型的H型CO2电还原系统
3.2 连续流电解池
通过在气室和液室之间使用气体扩散电极(gas diffusion electrode,GDE),可以缓解催化剂表面的气液界面物质传输问题。Hara 等采用GDE组装了Co、Ni、Rh、Pt、Pd、Cu及Ag电极,用于CO电还原产合成气,CO气体压力小于50atm(1atm=101325Pa)。在Ag-GDE电极体系中,当CO压力为20atm时,CO产物法拉第效率达86%,电流密度高达300mA/cm;当压力大于30atm 时,电流密度高达3.05A/cm。而对于Pd-GDE 电极,电流密度从0~1000mA/cm,CO 和H比例在4~1 之间。Xiang 等采用原位电化学沉积的方法首次制备了GDE表面修饰的Cu-In电催化剂。这种核壳结构的Cu-In催化剂电还原CO时产物CO法拉第效率高达90%以上,当电解电位(.RHE)为-1.17V时,电流密度高达200mA/cm以上,使得2.0cm电极表面上CO 产率可达3.05mg/min(CO 流速为15mL/min)[图8(a)、(b)]。
为解决H型电解池中,由于传质受限而导致的电流密度偏小问题,研究者们在气体扩散电极的基础上开发了连续流反应器。Xie 等制备了Bi-Pd单原子合金(single atom alloy,SAA)纳米枝晶(nanodendrites,ND)催化剂,其中Bi 原子单分散在Pd矩阵中,用于CO电还原产CO 反应中。在以BiPd-SAA ND为催化剂时,对比了H型电解池和连续流电解池,其CO 法拉第效率分别为90.5%和91.8%,过电位分别为290mV和200mV[图8(c)~(e)],在连续流反应器中,电流密度高达173mA/cm,远高于H型电解池(1.9mA/cm)。

图8 合金催化剂电还原CO2
3.3 固体氧化物电解池
固体氧化物电解池(solid oxide electrolysis cell,SOEC)可以将CO和水转化为合成气及烃类燃料等。电解池具有全固态和模块化结构、能量效率高、成本低等优点,在CO转化和可再生清洁电能存储方面极具应用潜力(图9)。钙钛矿型陶瓷阴极由于在氧化还原气氛下结构稳定,且可有效抑制积炭反应,是近年来SOEC 领域的研究热点。然而,钙钛矿型陶瓷阴极氧空位浓度低,CO吸附弱,CO活化和转化困难,导致CO电催化还原性能较低。Zhou等制备了钒掺杂的镧锶铁与钆掺杂的氧化铈纳米复合材料(LSFV/GDC),作为SOEC阴极应用于高温CO电催化还原反应。实验和理论计算结果表明,钒的掺杂可增加阴极氧空位浓度,提高了阴极CO高温吸附活化能力和电催化还原性能。在800℃、1.6V 时,SOEC 电流密度可达0.62A/cm,比未掺杂时提高了51.2%,法拉第电流效率接近100%。该研究通过金属元素掺杂来调控SOEC阴极材料氧空位浓度和CO吸附活化能力,为提高SOEC 阴极CO电催化还原性能提供了新思路。Gaudillere 等研究了Cu 基SOEC 电解池,用来电解CO和HO 产合成气,反应温度为600~700℃。由结果可知,SOEC 体系中,在中温区电解CO和HO产合成气是可行的。

图9 高温CO2电解的碳循环过程[79]
SOEC 电解CO的成本在很大程度上取决于耗电量,电力成本占SOEC 电解CO总成本的80%以上。因此,开发高性能和稳定的SOEC,降低电力消耗,对提高其经济竞争力至关重要。为此,SOEC 电还原CO技术未来的研究主要集中在以下几个方面:①基于DFT 计算,利用原位表征技术监测电极反应的基本过程和SOEC的衰减过程,为进一步探索高活性电极材料提供指导;②通过调节双钙钛矿氧化物、相结构以及引入活性界面,增强材料性能进而促进电化学反应;③通过将甲烷氧化偶联、低碳烷烃氧化脱氢、甲烷芳构化或其他化学反应耦合到SOEC 阳极,实现SOEC 多功能化,不仅可以促进阴极CO电化学还原,而且能够充分利用阳极产生的氧物种实现烷烃高效转化;④降低SOEC系统内部阻抗,提高系统效率等。
3.4 膜反应器电解池
由于H型电解池在应用过程中存在槽压高、液态产物有效浓度低等问题,因此,研究者们开发设计了基于质子交换膜燃料电池(proton exchange membrane fuel cell,PEMFC)的膜电极构型的电解池,其具有易于模块化且可规模化实现CO电催化转化的优势,应用潜力巨大。采用阴离子交换膜电极组件(anion membrane electrode assembly,AMEA)可以使电流密度极大提高。Jiang 等研究了氮掺杂石墨烯负载Ni 单原子催化剂(Ni-NG)电化学还原CO产合成气的催化活性。当采用膜电极组件时,电流密度可高达50mA/cm,Ni-NG 能够连续反应8h。在实际应用中,通过增大气体扩散电极(GDL)尺寸、增加催化剂载量以及增多膜组件数量等方式,能够增强AMEA膜电极电化学池转化效率。
在基于膜电极的电解池中,当使用阳离子交换膜(CEM)时,阳极必须在酸性介质中,需要使用贵金属OER 催化剂,增加成本,且易使阴极发生析氢反应,当使用阴离子交换膜(AEM)时,通常采用碳酸氢盐作为电解质,阴极产物易从阴极转移至阳极。通过将CEM 和AEM 同时层压在膜电极表面,制备成双极膜(BPM),能够显著改善上述问题。基于BPM的电解槽可以通过将H和OH离子分别选择性地传输到阴极和阳极来维持两侧的恒定pH,增强催化反应活性及电解池稳定性。Li 等研究了基于双极膜组件的电化学反应池中,采用非贵金属催化剂电还原CO产合成气,电流密度可高达200mA/cm。在基于双极膜组件的电解池中,能够实现对质子的有效管理,从而能够使系统稳定运行,并且在高电流密度下实现CO的电催化反应(图10)。
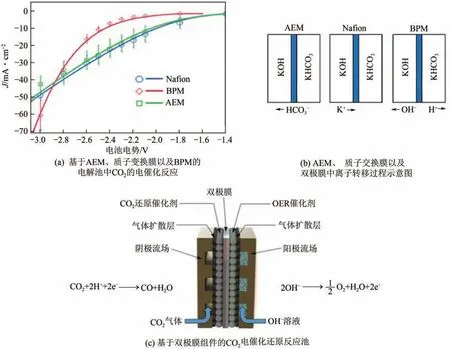
图10 基于双极膜电解池的CO2电还原技术[89]
4 产合成气效率提升策略
4.1 阳极反应耦合
典型地,电化学CO还原产合成气体系包含阴极的CORR、HER 和阳极的OER 过程。电化学还原CO过程中,电子从阴极电极表面转移到溶液中的CO分子或CO溶剂化离子中。由于将电子提供给CORR 的阳极OER 反应动力学过程缓慢,根据Hess 定律,反应体系70%的能量消耗都是来自OER过程,因此以OER作为阳极反应会导致CO电催化体系较大的过电势和较高的总能量消耗。由于多数的电催化CO还原产合成气体系都只考虑了阴极半反应,而没有考虑体系整体的电流效率、过电势和能量效率。因此,在阳极室以热力学上有利的反应(醇氧化、尿素氧化等)来代替阳极OER,能够增强电解池整体的能量效率,降低槽压。Verma 等通过共电解CO和丙三醇,采用丙三醇氧化反应代替OER时,可使电能消耗降低53%,生成产物乙二醇和乙醇。Li 等通过在Cu-In 电极上电还原CO产CO,电流密度为3.7mA/cm,CO法拉第效率大于70%;同时阳极铂电极表面采用醇氧化代替OER 反应,可生成比氧气更具有实际应用价值的产物,产率>75%[图11(a)]。
特别地,使用合适的电解质和活性催化剂,在同一电化学反应器中同时从阳极和阴极产生相同的产物更具有应用前景。Wei 等以过硫酸铵为氧化剂,在氢氧化钠溶液中一步氧化法制备了泡沫铜表面负载氧化铜纳米片(CuONS/CF),并在阳极上电催化部分醇氧化(MOR)为甲酸;同时合成了碳布上生长的介孔二氧化锡(mSnO/CC),并将其作为电化学还原CO的高效阴极催化剂,生成甲酸。在阳极上用动力学上有利的部分MOR 取代OER,可以显著降低阳极和电池的电位输入。在甲醇电解液中使用CuONS/CF 作为阳极催化剂,电流密度达到10mV/cm时的电位比OER 降低250mV,法拉第效率达到95%[图11(b)]。此外,mSnO/CC具有较高的催化活性,电催化CO还原产甲酸的法拉第效率可达81%。当阳极电极为CuONS/CF、阴极电极为mSnO/CC 时,所构建的电解槽仅需0.93V 的槽电压即可达到电流密度10mA/cm,相比相同催化条件下,电池电压降低500mV。

图11 阳极反应耦合阴极CO2还原反应
4.2 双活性位催化剂结构设计
电催化CO还原产合成气反应过程中,催化剂表面同时发生着CORR 及HER 两个反应过程,CORR 过程产生COOH、CO 等中间产物,HER 过程会形成H 等中间物质,根据Sabatier 原理及以上分析,中间态物质与催化剂表面结合(化学吸附)既不太强也不太弱时,才会具有最佳的催化活性。因此,通过调节催化剂固有电子结构,获得其表面对COOH、CO、HO 及H 等中间产物最佳的化学吸附特性,调控CO和H组成比例,实现高效的CO电化学还原产合成气反应催化活性。
通过双活性中心协同调控催化剂表面对合成气中间产物适宜的吸附能,对提高电还原CO产合成气反应的催化活性具有重要意义。He 等利用葡萄糖、双氰胺以及过渡金属盐溶液混合物作为前体,制得Co、Ni 双金属单原子催化剂,当电解电位为-0.5~-1.0V(RHE)时,CO/H比例为0.23~2.26,理论计算结果表明,Ni SAs 为产CO 活性中心,而Co SAs 是产H的活性中心。Song 等通过热解ZnO@Zn/Co-ZIF 纳米球前体,利用过量Zn 的挥发,抑制热解过程中Co原子团聚,获得了Co质量分数高达3.4%的Co-CN单原子催化剂,其Co原子表面电子结构有利于吸附CO分子,能够拉长C=O双键,活化CO分子,是CORR的活性中心,催化剂中吡啶氮和石墨氮等含氮官能团则是HER反应活性中心。在双活性位催化剂中通过调控双活性位点的比例来调节合成气组成比例。
另外,根据文献报道,过渡金属单原子催化剂具有较高的催化CO还原生成CO活性,而Fe、Co、Ni 等金属纳米颗粒则具有较好的析氢活性。Jiang 等研究对比了氮掺杂石墨烯负载Ni 单原子催化剂(Ni-NG)与石墨烯负载Ni 纳米颗粒(Ni-NPs/G)电催化还原CO活性,结果表明Ni 纳米颗粒几乎只有析氢活性,而Ni 单原子还原CO时,CO 选择性高达97%。基于此,Daiyan 等构建了钴单原子协同纳米颗粒(Co@CoNC-900)的双活性位型催化剂,通过Co 单原子与纳米颗粒间相互作用,调控催化剂固有电子结构,从而得到组成比例可调的合成气产物,实现了高效电催化CO还原。
为了构建过渡金属双活性位型催化剂,有研究者采用沸石咪唑酯骨架材料(zeolite imidazole frameworks,ZIFs)为前体/模板,利用ZIF 的孔道限域及配位键合等方式固定金属原子。因ZIFs 具有丰富的氮源和可调控的孔道结构,其有机单体对金属的锚定作用以及笼状ZIFs 对内部金属的封装效果都有利于限制金属原子在高温炭化过程中的迁移与团聚,这对于形成高分散的金属活性位点有极大的帮助,通过调控热解过程实现金属单原子(single atoms,SAs)协同纳米颗粒(nanoparticles,NPs)双活性位型催化剂的构筑。通过构建过渡金属NPs/SAs 双活性位型催化体系,协同调控CORR和HER 催化活性,从而获得可调比例的合成气产物。
4.3 催化剂多级形貌调控
针对CO电还原产合成气反应过程中三相界面处的传质问题,通过采取分级形貌结构调控,可获得更高密度的、均匀分散的活性位点以及互相连通的导电框架,实现电催化剂表面传质过程的强化。研究者多通过零维(0D)碳纳米球、一维(1D)碳纳米管和二维(2D)石墨烯等碳材料相互作用,巧妙构建多级结构框架作为碳载体,以增强反应催化效率。Sun等利用MOF媒介拓扑转化策略在泡沫镍(NF)上构建了多孔的三元CoFeNi LDHs 框架,其孔洞边缘部分含有丰富的缺陷位及未配位的金属活性中心[图12(a)]。这种多级结构的形成增大了催化剂比表面积,获得更多的活性位点。Yan等采用模板法在碳纳米棒上原位生长了Ni纳米颗粒和氮掺杂碳纳米管,形成了Ni@N-CNT/NRs分枝状的多级结构,具有丰富且分散均匀的活性位点、较大的比表面积、较小的电荷转移和物质扩散阻力以及较强的机械稳定性[图12(b)]。Pan 等采用氧化开壁的方法使多壁碳纳米管部分开壁,形成紧密结合的碳纳米管/纳米带复合结构,最终制备了Fe、N共掺杂的Fe-N/CNT@GNR框架[图12(c)],因其较高的电化学比表面积、较强的物质转移能力及丰富的多孔结构,使其具有高电催化CO还原活性,CO法拉第效率可达96%,电流密度为22.6mA/cm。

图12 碳多级结构
碳纳米管是一种中空管状的一维(1D)碳结构材料,电子转移能力强,石墨烯作为一种具有超大比表面积及超强导电能力的二维(2D)材料,也备受学者青睐,碳纳米管和石墨烯作为单原子载体已有较多的研究报道。有研究者结合石墨烯和碳纳米管的结构特性,构筑了类似三明治夹心的多级(1D/2D)碳结构负载过渡金属电催化剂,具有以下几方面的优势:①可提高催化剂比表面积,增加活性位点密度;②碳纳米管和石墨烯超强的导电性,可增强催化剂电子转移能力,促进CO电还原反应;③三明治夹心的多级碳结构,有助于形成丰富的孔结构,强化气、液传质过程;④可避免多孔碳杂乱堆叠和导电性差的问题,同时碳纳米管、石墨烯之间的界面结构对于金属活性位的形成以及催化活性的提高也有重要意义;⑤有助于提高催化剂稳定性。
5 结语和展望
电化学还原二氧化碳(CORR)利用可再生能源,将CO资源化,生成高价值的燃料和化学品,是有助于实现全球可持续能源经济的一项有前途的策略,但在实现低成本、大规模商用之前,仍有众多问题值得关注。
第一,亟需开发新的方法来加快高活性催化剂的设计和筛选。新兴的机器学习和高通量实验方法的发展为催化剂预测筛选能力提供了解决途径,有助于材料发展和CORR电催化剂的设计。
第二,现有理论基础对于描述电化学界面电荷的迁移,以及在反应条件下评估反应动力学和反应能垒方面仍然存在挑战。多尺度模拟有助于更好地了解真实反应中的界面过程,是增强实验和理论研究相融合的有效手段。
第三,结合原位技术,包括原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)、原位表面增强拉曼散射、原位同步辐射、原位X射线光谱等技术深入探究反应中间产物、反应机理等。
第四,开发低成本、高性能催化剂,增大电流密度,增强催化活性及选择性,提高催化剂稳定性。
