二氧化碳与不饱和烃制备丙烯酸及其衍生物研究进展
2022-04-12岳成光姬文豪冯帮满王美岩马新宾
岳成光,姬文豪,冯帮满,王美岩,马新宾
(天津大学化工学院,绿色合成与转化教育部重点实验室,天津 300350)
“力争2030年前实现碳达峰、2060年前实现碳中和”的“双碳”目标是中国基于推动构建人类命运共同体的责任担当和实现可持续发展的内在要求作出的重大战略决策。“碳达峰、碳中和”目标的实现,需要各行业努力协作地加快关键核心技术突破和经济结构转型升级。化工行业作为低碳经济的重要支柱,理应担当起节能减排、调整优化产业结构与能源结构的科技攻关重任。而绿色低碳化工体系的建立需要实现原料低碳化、过程低碳化、末端低碳化以及CO捕集与利用。其中CO捕集与利用是减少CO排放和保障能源安全的重要战略技术选择之一。从资源化利用的角度,CO是一种廉价易得的C资源和氧资源,将其作为原料通过化工转化制备高附加值化学品,可将CO变“废”为“宝”,实现CO的固定。但是CO整个分子处于最低能量状态,具有热力学稳定性和动力学惰性,因此CO的化工转化一般需要比较苛刻的反应条件(如高温、高压),且CO资源化利用的范围也受到很大限制。因此,开发新型高效的CO转化利用技术具有很高的社会经济价值和生态意义。
丙烯酸(酯)是工业上十分重要的有机合成原料,其自聚或与其他单体如丙烯腈、丁二烯、氯乙烯、顺酐等发生共聚,可生产合成树脂、合成纤维、合成橡胶、涂料等广泛应用于建筑、纺织、塑料加工、包装材料等领域的大宗化工原料,因此其合成方法的开发与优化具有重要意义。目前已有的丙烯酸(酯)主要合成方法有丙烯氧化法、丙烯腈水解法、乙烯酮法等,其中丙烯腈水解法和乙烯酮法由于污染环境、原料成本高、中间产物有致癌性等问题被逐渐淘汰。丙烯氧化法由于原料廉价易得、工艺可行性高,成为目前工业生产丙烯酸(酯)的主流工艺,但是该工艺的物料消耗和能耗还有待进一步优化(见表1)。

表1 丙烯酸(酯)主要合成工艺对比
以CO为羧基源与不饱和烃发生羧化偶联反应,是一种新型、绿色、具有应用潜力的丙烯酸及其衍生物合成方法,同时也是实现CO高值化利用的有效途径。根据反应原料和机理的不同,该工艺主要有三条路径:CO与烯烃的偶联羧化、CO与炔烃的还原羧化以及CO与联烯的还原羧化(图1)。本文将详细介绍近年来以CO为羧基源合成丙烯酸及其衍生物的研究进展,着重描述在上述三条路线中不同金属催化剂的催化特点及其作用机制。

图1 CO2为羧基源的丙烯酸及其衍生物合成路线
1 CO2与烯烃偶联羧化
1.1 镍催化CO2与烯烃偶联羧化
19 世纪80 年代,Hoberg 等发现CO与烯烃可在金属络合物(LM)上发生偶联羧化反应,生成金属杂环内酯。当Ni作为金属络合中心时,乙烯、CO与Ni 络合物形成的镍内酯可以水解得到丙酸[图2(a)]。当使用苯乙烯代替乙烯参与上述反应时,水解温度为60℃得到的产物为饱和羧酸(苯丙酸);水解温度为85℃生成的产物为,-不饱和羧酸(肉桂酸)且收率能够达到70%[图2(b)]。可以看出较高的水解温度能够促进镍内酯中-H 的消除,这也说明-H 的消除是产物形成不饱和碳碳双键的关键因素。因此,研究者们设想如果能够通过条件优化,实现乙烯镍内酯的-H 消除,将有可能经由CO与乙烯制备得到丙烯酸。此外,上述偶联羧化反应需要化学计量的Ni(DBU)(DBU为1,8-二氮杂双环[5.4.0]十一碳-7-烯)促进,且在反应结束后,体系中的Ni(DBU)无法再生。Hoberg等认为反应过程中-H 消除后会由Ni 向配体DBU 转移,使得镍络合物失活。

图2 Ni(0)促进CO2与乙烯偶联羧化反应与Ni(0)促进CO2与苯乙烯偶联羧化反应
2006年,Fischer等发现可以通过调控镍络合物的配体类型来促进镍内酯的-H 消除。其中,双二苯基膦类配体[如双(二苯膦基)甲烷,dppm]对镍内酯-H 消除的促进作用最佳(图3)。但是,镍内酯经-H 消除后,容易形成稳定的Ni-Ni二聚物,使得丙烯酸无法顺利脱离。

图3 双二苯基膦类配体-Ni促进的CO2与乙烯的反应路径图[12]
Graham等利用密度泛函理论(DFT)对Hoberg等提出的丙烯酸生成路径以及催化剂失活路径进行了理论计算。计算发现,Ni催化CO与烯烃生产丙烯酸路经可分为三个部分(图4),其中-H 消除所需能量最高,这一结果与实验结果相一致。对催化剂失活反应的研究表明,H 从Ni 中心转移到DBU,必须克服150.2kJ/mol 的能垒,而且生成的产物相对于初始反应物的能量为+102.6kJ/mol,从而排除了该氢转移路径是催化剂失活的原因。他们认为整体反应热力学是导致该催化反应无法顺利进行的主要原因。此外,DFT计算结果表明,镍氧五元杂环中Ni—O的延长能够在一定程度上减弱环的应力作用,利于-H的消除。

图4 Ni(DBU)2促进CO2与乙烯偶联羧化反应的主要反应步骤及其过渡中间体的相对能量[13]
基于上述发现,研究者们设想通过引入开环剂诱导Ni—O 断裂,进而促进后续反应的进行。Bruckmeier 等采用碘甲烷(MeI)对镍氧五元杂环进行原位甲基化处理,促进了Ni—O的断裂,首次成功地从镍内酯中解离得到丙烯酸甲酯,产率为33%。虽然该研究最终没能实现催化循环,但是其通过原位甲基化策略突破了CO与乙烯偶联羧化反应过程中较具挑战的两步:实现了-H 的消除以及丙烯酸酯的释放(图5)。这些发现对于后续研究具有十分重要的参考价值与指导意义。

图5 MeI参与的Ni(0)催化CO2与乙烯偶联羧化机理[14]
2011 年,Lee 等在Bruckmeier 等工 作的基础上较为系统地考察了不同配体对反应结果的影响。结果表明,,,′,′-四甲基乙二胺、双(二苯基膦基)乙烷、双(二苯基膦基)丙烷作为配体时,对应的镍内酯能够开环生成丙烯酸甲酯;而当双(二苯基膦基)丁烷和吡啶作为配体时,对应的镍内酯不发生反应。这一现象可能与不同配体的电子和空间效应不同有关。此外,他们认为MeI参与的A物种的开环过程是关键反应步骤(图6),增加MeI的比例可促进该步骤的正向进行,进而提高丙烯酸甲酯产率。

图6 MeI参与的镍内酯开环反应步骤及其理论计算能垒[15]
2012 年,Lejkowski 等首次 实现了Ni(0)催化CO和乙烯偶联羧化的催化循环,发现整个催化反应循环大致可以分为三个步骤:镍内酯形成、开环以及后续的丙烯酸钠脱去。Lejkowski 等将这三个催化步骤独立研究,分别考察了配体结构、溶剂、反应物分压、开环剂等因素对反应体系的影响。最终使用二叔丁基膦乙烷(dtbpe)作为Ni(0)的配体,引入叔丁醇钠(BuONa)作为开环剂,用CO和乙烯合成出丙烯酸钠,产率达到90%。他们发现该催化体系中镍内酯的形成过程需要在高分压CO下进行(图7,过程A);但对于镍内酯开环过程B,高分压CO会导致CO与碱生成副产物碳酸酯而消耗大量的BuONa,因此开环过程B需要在低分压CO下进行以降低副反应发生概率;最后丙烯酸钠的脱去在高分压乙烯条件下更易发生。这就使得整个催化循环需要在不同反应条件下进行,从而增加了丙烯酸钠合成工艺的复杂性,而且催化剂的周转数较低(TON=10)。因此寻找BuONa 的合适替代品,使其能够在体系中与CO共存而不发生副反应,应该是提高TON的有效策略之一。

图7 Ni(dtbpe)催化CO2和乙烯偶联羧化反应机理图[16]
2013 年,Dong 等发现三(五氟苯基)硼烷能够快速促进1,1-双(二苯基膦基)二茂铁(dppf)镍内酯的开环以及-H消除。与之前研究不同的是,镍内酯开环后的热力学产物为(dppf)Ni(CH(CH)COBAr)。这表明-H消除后的瞬态产物不稳定,会经由2,1-插入生成镍氧四元杂环。该物质在叔丁基氨三吡啶膦(BTPP)或DBU 等有机碱作用下能够得到丙烯酸盐-Ni(Ⅱ)的络合物,但不能发生进一步的解离得到丙烯酸盐(图8)。虽然该研究未能实现整个反应的催化循环,但是他们将Lewis 酸引入的策略很好地促进了镍内酯络合物的开环,为叔丁醇钠这类Brϕnsted碱型开环剂找到了合适的替代品,理论上能够避免开环剂与CO发生副反应。
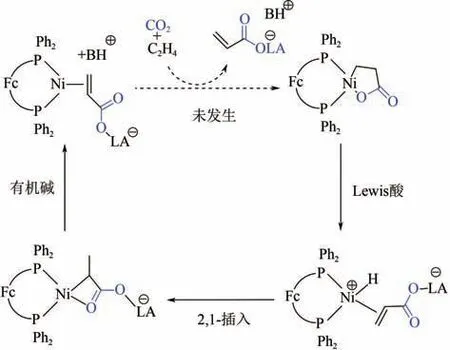
图8 Lewis酸为开环剂参与的Ni(0)催化CO2与乙烯偶联羧化反应路径[17]
随后,Hendriksen 等以LiI 作为Lewis 酸,三乙基胺作为碱,来促进镍内酯的开环以及-H 消除,实现了CO与乙烯的偶联羧化合成丙烯酸锂(图9)。他们考察了不同双膦配体结构对催化反应的影响,发现1,3-双(二环己基膦)乙烷(dcpe)作为配体时,反应结束后混合物会变为紫色,说明生成了无催化活性的[(dcpe)NiI][Ni(Ⅱ)]副产物,为了将Ni(Ⅱ)副产物还原,体系中需要额外加入锌粉作为还原剂来实现整个催化反应循环。而1,3-双(二环己基膦)丙烷(dcpp)作为配体时,改变了Ni 络合物中螯合环的大小和金属配位角,使得上述类似的Ni(Ⅱ)副产物的生成被抑制,此时无需还原剂的加入,整个催化反应循环也能顺利进行。此外,他们还考察了反应体系中乙烯和CO的分压对反应的影响,发现通过增加乙烯/CO的分压比能促进丙烯酸钠的脱去从而提高TON。最终他们通过优化反应条件,在无锌粉加入情况下使得整个催化体系的TON 达到16;如果将催化剂浓度减半并额外加入锌粉作为还原剂,TON能够提高到21。

图9 LiI参与的Ni(0)催化CO2与乙烯偶联羧化反应[18]
同年,Huguet等在已有研究的基础上对该反应体系中的开环剂、配体、温度、压力、溶剂等因素进行了系统的考察。他们发现:2-氟苯酚钠具有碱性强且弱亲核性的性质,既能实现-H消除,又能与CO共存从而避免自身发生副反应而被消耗。在使用2-氟苯酚钠作为开环剂,(,’)-(+)-1,2-双(叔丁基甲基膦基)苯(BenzP)作为配体,THF为溶剂,CO分压为20bar(1bar=10Pa)、CH分压为10bar,反应温度100°C,采用锌粉为还原剂时,该催化体系的TON能够达到107,产物为丙烯酸钠。
经过不断探索与创新,Ni催化烯烃与CO羧化加成反应体系有了长足的发展,实现了由化学计量反应向催化反应的跨越。而且在此过程中研究者们作出很多富有创意的尝试,这些进步不仅促进了该催化体系的性能提升,而且相关的思路和方法也值得其他类似体系的研究者们参考。在此,本文对上述多种Ni促进的乙烯与CO羧化加成反应体系进行了归纳总结,并对比了几种实现Ni 催化的乙烯与CO羧化加成的反应体系(见表2)。可以看到,在Lejkowski 等开创性地以两步法实现Ni(0)催化CO和乙烯偶联羧化的催化循环后,Hendriksen和Huguet等在此基础上对该反应体系中的开环剂、配体、还原剂、温度、压力、溶剂等因素进行进一步优化,并分别实现了该催化体系的一锅法反应以及TON 的显著提升,他们的努力使得该催化体系有了工业放大的可能性。

表2 Ni催化乙烯与CO2羧化加成反应体系对比
2015 年,Jevtovikj 等报道了镍络合物能够催化乙烯以外的烯烃生成丙烯酸的衍生物,-不饱和羧酸盐。而且他们对反应路径提出了更加详细的分析:在镍络合物的催化下,含有取代基的烯烃可以通过镍-烯烃络合物的形式与CO发生羧化反应生成镍内酯络合物,然后离解得到镍-不饱和羧酸盐络合物,乙烯与不饱和羧酸盐替换完成催化循环并生成,-不饱和羧酸盐(图10)。与乙烯作为底物时不同的是,取代烯烃作为底物时,镍内酯的形成是整个催化反应的关键步骤。
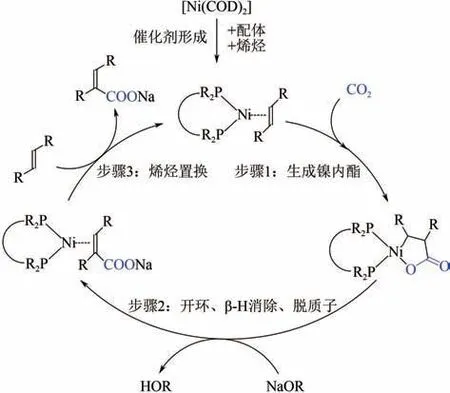
图10 Ni催化取代烯烃与CO2羧化反应生成α,β-不饱和羧酸盐的反应机理图[20]
1.2 钯催化CO2与烯烃偶联羧化
Pd与Ni性质相似,也能形成钯氧五元杂环中间体。Stieber等采用(COD)PdCl/1,2-双(二环己基膦基)乙烷(dcpe)为催化剂、2-氟苯酚钠为开环试剂,研究了Pd(0)催化CO与乙烯的偶联羧化反应(图11)。反应需加入锌粉作为还原剂,且TON仅有29,远小于对应的镍络合物催化剂。研究者发现反应过程中2-氟苯酚钠与CO生成的羧酸盐与关键中间体钯内酯会发生副反应生成无催化活性的Pd(Ⅱ)络合物,这可能是该催化体系效率不高的原因。
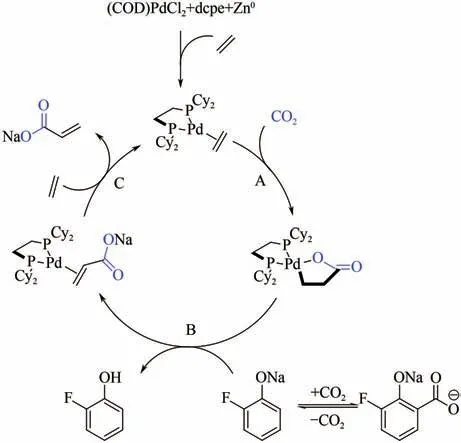
图11 (COD)PdCl2催化CO2与乙烯的偶联羧化反应机理图[21]
Manzini 等在Stieber 等研究基础上,将反应溶剂由四氢呋喃改为苯甲醚,使用高亲脂性的2,6-二叔丁基-4-甲基苯酚钠替换2-氟苯酚钠,在无需额外添加Zn 粉作还原剂的条件下,催化剂的TON达到了106。研究者认为活性的提高与丙烯酸钠和2,6-二叔丁基-4-甲基苯酚在苯甲醚中溶解度低有关。结合上述Stieber等的研究发现,认为较低的产物浓度促进了主反应的正向进行而降低了副反应发生的概率,从而提高了催化反应效率。值得注意的是,他们使用Pd(PPh)[Pd(0)前体]代替了(COD)PdCl[Pd(Ⅱ)前体],因此体系中无需额外加入锌粉作为还原剂。这一改进不仅简化了丙烯酸钠的生产工艺,还降低了其生产成本。
为了满足工业化放大的需求,Manzini等对以上催化体系作了进一步优化。他们发现以[Pd(PPh)/dcpe]为催化剂时,采用酰胺作为溶剂能明显提高反应TON,推测酰胺作为溶剂具有较大CO溶解度,而高CO浓度利于稳定活性物质促进钯内酯的形成并抑制低聚物的形成,从而提高反应的TON。当使用-环己基吡咯烷酮(CHB)作为溶剂时,TON最高达到514。更重要的是,由于使用了酰胺溶剂,催化剂能够直接经由相分离回收循环而不需要额外的再生步骤(图12)。此外,将叔丁醇钠或异丙醇钠作为碱加到体系中,由于其对应醇的沸点较低,因此可以采用分馏加碱化的方式进行循环。这大大简化了其化工过程并降低了其能耗与生产成本,使得CO与乙烯合成丙烯酸的工艺向工业放大迈出了重要的一步。

图12 Pd催化CO2与乙烯合成丙烯酸钠的工艺概念图[23]
可以看到,Ni催化CO与乙烯偶联羧化反应的相关研究虽然未能实现工业化,但是其对于该反应体系相关机理的探究具有很大价值。研究者们为了打通反应循环中镍内酯的开环以及-H消除过程,进行了包括优化配体、更换开环剂等很多尝试,这些研究发现也为后续Pd 催化剂的性能改善提供了很好的理论支持。就目前来看,Pd 催化剂在该体系中表现出较优的催化性能,已有研究者开发出对应的催化工艺流程,这为实现Pd 催化CO与乙烯偶联羧化反应制备丙烯酸类衍生物的工业化迈出重要一步。
除了Ni、Pd类金属催化剂,Mo/W、Fe、Pt、Rh等金属络合物催化的CO与烯烃偶联羧化反应也得到研究,但催化效果均不理想,在此不作详细介绍。
2 CO2与炔烃的还原羧化
炔烃相对于烯烃更活泼,理论上CO与炔烃的羧化反应更易发生,因此CO与炔烃还原羧化制备丙烯酸衍生物的研究一直备受关注。与烯烃的偶联羧化类似,镍络合物也是CO与炔烃反应制备丙烯酸衍生物体系的主要催化剂。炔烃本身具有2个不饱和度,生成的镍内酯无需经历-H 消除即可得到丙烯酸或其衍生物。且考虑到炔烃结构的可修饰性,该反应更适用于含有取代基的高附加值丙烯酸衍生物的制备。
2.1 镍催化CO2与炔烃还原羧化
1982 年,Hoberg 等发现将CO鼓泡通入含有2-丁炔、Ni(CDT)和四甲基乙二胺的混合物时,能够生成五元镍内酯(产率为65%)。镍内酯经由酸解后可以得到二甲基-2丁烯酸(图13)。由于炔烃更好的反应活性,该反应无需加压或引入开环剂。这明显改善了CO基丙烯酸衍生物的制备条件。

图13 Ni(CDT)促进的CO2与2-丁炔还原羧化反应[37]
对于该反应路线,非对称炔烃的产物会有多种同分异构体,如何提高产物的区域选择性成为一个挑战。1991年,Dérien等开发了一种使用Ni(bpy)(BF)为催化剂、(TBA)BF为电解质、DMF 为溶剂的电化学方法,用于促进CO与炔烃的还原羧化。在50mA恒流的单室碳/镁电池中能够实现端炔和内炔向,-不饱和羧酸的转化。对于端炔,烷基取代和芳基取代对其羧化率和区域选择性有明显影响。其中烷基取代的末端炔烃,其主要羧化产物为符合马氏规则的,-不饱和羧酸;而芳基取代的端炔,羧化产物中反马氏规则的,-不饱和羧酸比例明显提高。对于末端炔烃,CO羧化加成主要发生在空间位阻较小的碳上。
与Dérien等的研究结果不同,Saito等发现以DBU作为配体时,CO和端炔还原羧化的主要产物为反马氏规则的,-不饱和羧酸(图14)。他们认为不同的反应结果与配体有关:当使用DBU 作为配体时,Ni(DBU)与炔烃之间的作用更强,体系中镍活化炔烃物种f较为稳定且浓度较高,这促进了f与CO加成反应的正向进行,从而利于生成动力学有利的中间体g,其水解后的产物为反马氏规则的,-不饱和羧酸;而使用bpy作为配体时,Ni(bpy)与炔烃之间的作用较弱,这使得Ni(bpy)-炔烃物种b在体系中的浓度较低,因此动力学有利的中间体c形成后会由于逆反应的存在,无法参与进一步的水解反应,而热力学有利的中间体d形成后较为稳定,因此生成马氏规则的,-不饱和羧酸的速率更快。Graham 等通过DFT 计算也进一步证实了这种区域选择性受热力学和动力学控制。

图14 Dérien和Saito报道的Ni(0)促进的CO2与炔烃的还原羧化反应对比[39]
Takimoto 等通过在反应体系中引入有机锌试剂,能够在温和条件下实现炔烃还原羧化的同时引入另外一个有机基团,得到高区域选择性的,’-双取代的,-不饱和羧酸,如图15(a)所示。值得注意的是,当使用EtZn 作为有机锌试剂时,反应的主要产物是化合物Ⅵ′(没有发生乙基取代)。而使用其他有机锌试剂如BuZnI 时,即使其位上有H 原子,也不会发生-H 消除,可见EtZn 在该反应中具有一定的特殊性。设想机理如图15(b)所示,当R′为乙基时,Ⅲ′会首先发生-H 消除,脱掉一分子乙烯,得到中间体Ⅳ′,Ⅳ′发生还原消除得到锌盐Ⅴ′,Ⅴ′水解酸化后得到,-不饱和羧酸Ⅵ′。后来,Shimizu 等发现在体系中加入过量DBU,使其与烷基锌形成配合物,可有效促进转移金属化过程,进而提高产率。
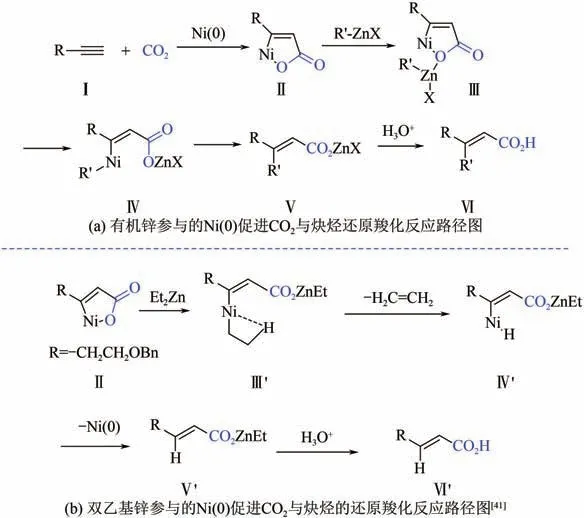
图15 有机锌参与的Ni(0)促进CO2与炔烃还原羧化反应路径图与双乙基锌参与的Ni(0)促进CO2与炔烃的还原羧化反应路径图[41]
Aoki 等基于DBU 配体设计了型双脒配体(图16),并研究了其对Ni 催化CO与炔烃还原羧化制丙烯酸衍生物反应的影响。他们发现配体结构是影响产物区域选择性的重要因素。当型双脒配体的R′是甲基或者丁基时,得到反马氏规则产物(>98%);而R′为H时,得到的是马氏规则产物。该研究通过调控配体结构能够让产物的区域选择性反转,为专一区域选择性丙烯酸衍生物的制备提供了可借鉴的方案。
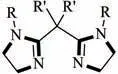
图16 endo型双脒配体结构[43]
2011 年,上海有机所麻生明院士团队设计了一条不经由镍氧五元杂环中间体的反应路径,首次实现了镍催化的CO与炔烃还原羧化反应。在Ni(COD)(物质的量分数1%~3%)催化下,EtZn先COD 与炔烃形成烯基锌,CO继而插入到被Zn取代的碳原子上,最后Ni 通过还原消除得以实现催化循环,同时得到产物丙烯酸锌盐(图17)。

图17 Ni(0)催化的CO2与炔烃还原羧化机理[44]
2015年,意大利Martin课题组发现使用醇代替敏感的有机锌作为质子源,可实现镍催化CO与芳基炔烃高区域选择性还原羧化,该路径条件温和、底物适用性广。由于芳基取代基的空间和电子效应,羧化产物几乎全是-苯基-,-不饱和酸,区域选择性专一(图18)。但是,反应需加入化学计量的Mn粉参与Ni物种的还原,以实现Ni(0)的再生循环。基于此方法,Saito等高选择性地得到了-取代--氨基丙烯酸,进一步对其不对称氢化能够得到-取代--氨基酸,这为手性-取代--氨基酸类物质的合成提供了一条新路径。

图18 醇作为质子源参与的Ni(0)催化CO2与芳炔羧化机理[45]
上述介绍的CO与炔烃还原羧化制丙烯酸衍生物研究中,对Ni 络合物结构、配体类型、添加剂等对反应活性及区域选择性的影响规律的考察对后续研究具有重要参考价值。尤其是还原剂的引入,促进了活性Ni物种的催化循环,为以CO和炔烃为原料制备丙烯酸及衍生物的进一步工业应用提供了研究基础。
2.2 其他金属催化CO2与炔烃还原羧化
Fujihara 等以(EtO)SiH 作为还原剂、含氮杂环卡宾为配体,首次使用IMesCuF 催化CO与炔烃的还原羧化反应,得到丙烯酸硅酯衍生物,经进一步酸化可得取代丙烯酸。该反应中CO插入烯基铜物种B 生成C 物种的过程需要较高的反应温度(65℃),而其他化学反应过程均能在室温条件下进行,因此可以判定该反应过程是整个催化反应的决速步骤(图19)。

图19 IMesCuF催化CO2与炔烃的还原羧化反应机理[46]
受此启发,Takimoto等利用三烷基铝(MeAl)或二烷基氢化铝(BuAlH)将不同炔烃烷基铝化或氢铝化得到链烯基铝化合物,然后使用氮杂卡宾铜IPrCuCl 催化其与CO羧化,得到,-不饱和羧酸。由于Cu 催化羧化反应中CO的插入是通过取代烯烃上的烷基铝物种实现的,所以整个反应的区域选择性仅由烷基铝化或氢铝化过程决定。而烷基铝化或氢铝化过程中的区域和立体选择性能够通过选择不同类型的催化剂实现,因此经由烷基铝化或氢铝化以及后续的羧化能够实现产物,-不饱和羧酸的构型调控。该团队还使用IPrCuCl 作为催化剂催化CO与炔酰胺的烷基化羧化反应,在温和条件下一锅法能够合成出不对称丙烯酸衍生物,-脱氢氨基酸。
2003 年,Six首次报道了Ti(OPr)促进的CO与炔烃的还原羧化反应,不过Ti(OPr)需要先在低温下与格氏试剂发生转金属化生成-环戊烯钛催化物种。与Ni(COD)催化机制类似,钛氧五元杂环是该过程的关键中间体,对其进一步酸化得到,-不饱和酸(图20)。随后,Shao等使用CpTiCl作为催化剂前驱体,也实现了CO与炔烃高效高选择性制,-不饱和羧酸。与Six 研究不相同的是,他们认为该催化反应的循环经炔烃加氢钛化、转金属化、CO羧化以及酸化水解四步完成,而没有形成钛氧杂环中间体(图21)。

图20 Ti(OiPr)4促进的CO2与炔烃还原羧化反应机理[49]
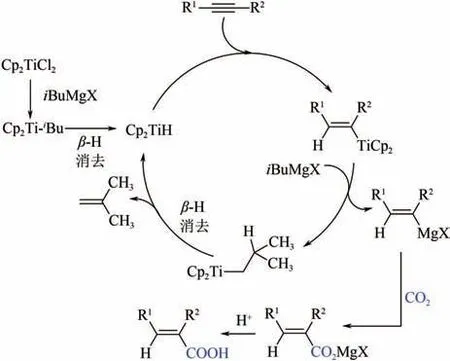
图21 Cp2TiCl2催化CO2与炔烃的还原羧化反应机理图[50]
与CuH物种类似,FeH物种也可催化CO与炔烃制丙烯酸类化合物的反应。Santhoshkumar等使用FeCl与EtMgBr 作用得到FeH 催化活性物种,经由与Cu 催化相似的炔烃加氢、CO插入、转金属化、酸化过程,最终得到产物,-不饱和羧酸。该方法使用无毒易得的FeCl作为催化剂前体,且反应条件温和,产物收率高,区域选择性专一。
此外,Co作为金属络合中心也可用于催化CO与炔烃的还原羧化反应。使用CoI(dppf)作为催化剂,加入锌粉作为还原剂时,该催化体系能经由钴氧五元环中间体得到-锌化丙烯酸盐,该物质中的烯基锌部分可以被替换为各种取代基,最终制备得到多取代的丙烯酸类化合物。
3 CO2与联烯的偶联羧化反应
联烯与炔烃碳碳三键的性质类似,由于其结构中含有两个连续不饱和键,理论上联烯能够参与两个分子的加成反应,这使得其与CO还原羧化产物的结构具有更高的可调控性。Takimoto 等报道了Ni(COD)促进的CO与联烯、芳基醛的三元偶联反应,能够制备得到-亚甲基--羟基羧酸。该Ni(COD)/DBU体系也适用于CO与三甲基硅基联烯羧化制备硅基取代的丙烯酸类衍生物。Tani 等以Cu 络合物为催化剂,实现了对上述CO与联烯硅羧化反应的催化循环。与Cu 催化的CO与炔烃还原羧化反应过程类似,C—Cu 键的形成及后续CO插入是反应的关键。另外,通过改变配体可实现反应区域选择性的调控,使用大位阻的Me-DuPhos 作为配体,产物为-硅烷基丙烯酸,而使用PCy配体则可得到-硅甲基丙烯酸衍生物(图22)。
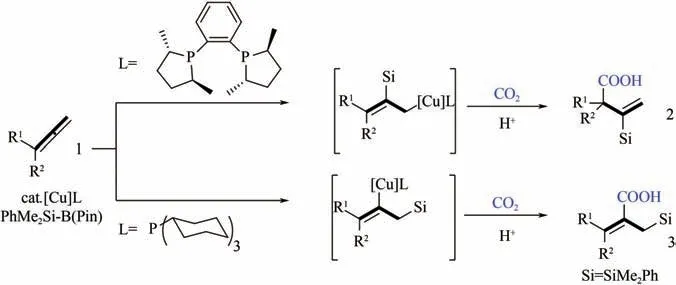
图22 不同配体对Cu络合物催化联烯硅羧化反应的影响[55]
过渡金属络合物催化不饱和烃(烯烃、炔烃和联烯)与CO羧化反应的相关研究已经发展了几十年,期间研究者们开发了多种过渡金属(Ni、Pd、Co、Ti、Cu、Fe 等)作为催化剂的活性中心。就目前研究结果来看,该反应体系主要有两种反应路径:①金属中心M与不饱和烃和CO形成内酯中间体(M),再经由开环、脱氢、置换等步骤生成目标产物;②金属配合物与不饱和烃形成M—C键得到链式中间体,然后CO再插入。Ni(0)、Pd(0)和Co(Ⅰ)在合适配体存在下都有与不饱和烃和CO形成五元金属内酯[Ni(Ⅱ)、Pd(Ⅱ)和Co(Ⅲ)]的能力,而后续该中间体开环的难易程度对其催化性能有着决定性的影响。对于具有多种氧化态的Ti,就目前研究结果来看,Ti(Ⅱ)物种能够生成钛内酯[Ti(Ⅳ)],Ti(Ⅲ)物种则倾向于先与炔烃形成烯基钛化合物中间体,再发生二氧化碳插入。对于Cu(Ⅰ)和Fe(Ⅱ)这类催化中心,由于无法再失去两个电子,因此无法与不饱和烃和CO形成内酯,发生的是与Ti(Ⅲ)物种类似的反应路径。因此,本文作者推测在该体系中,催化剂金属中心的电子得失能力对催化反应路径存在一定的影响。
此外,不同结构的不饱和烃,与CO羧化后的产物结构也有着较大区别。烯烃与CO羧化体系中乙烯与CO羧化加成制备丙烯酸是目前较有应用前景的工艺路线。如果该工艺能够实现工业放大,将会在产生巨大的经济效益的同时实现CO高值化转化,助力国家“双碳”目标的早日实现。对于炔烃以及联烯与CO的羧化体系,由于产物存在区域与立体选择性,在获得高催化活性的同时,如何提高产物的选择性将成为日后研究的重点。如果能够经由该路径实现不饱和烃向高区域选择性羧化产物的高效转化,那么对于相关精细化学品(如药物中间体等)合成路径的优化将起到积极的作用。
4 结语
本文详细介绍了CO与不饱和烃为原料通过羧化反应制备丙烯酸及衍生物的研究进展。该路线的开发不仅为丙烯酸基聚酯材料的合成提供了一条绿色可持续的原料生产路线,也为许多高附加值精细化学品的获取提供了一种很好的制备方法。目前CO与不饱和烃制备丙烯酸的催化体系仍以金属Ni催化为主。对于CO与烯烃的偶联羧化路线,镍氧五元内酯中间体的形成及-H 消除是反应进行的关键步骤和难点,而Ni(0)、Pd(0)催化物种的稳定性及循环再生也是该路线需进一步突破的关键科学问题。炔烃和联烯为原料时,虽无需经历-H 消除过程,但同样存在催化剂的循环问题。随着研究的不断深入,一些性能优异的金属催化剂也不断被发掘,如Pd、Cu 催化剂等,但在催化活性、稳定性、反应参数等方面仍需进一步探索。总体而言,以CO和不饱和烃为原料制备丙烯酸及衍生物的研究虽已实现了由化学计量反应向催化反应的跨越,但仍处于实验室研发阶段。相信随着催化机理的深入探究、催化剂设计与优化等研究工作的不断推进,该合成路线有望实现工业应用,助力绿色低碳化工产业体系的建立以及我国“双碳”目标的顺利实现。
