甘草炭炮制工艺及质量标准研究△
2022-04-11李本淳王婧姝李岩商丽丽赵艳
李本淳,王婧姝,李岩,商丽丽,赵艳*
1.四平市食品药品检验所,吉林 四平 136000;2.吉林师范大学,吉林 四平 136000;3.白城市食品药品检验所,吉林 白城 137000
甘草炭为豆科植物甘草、胀果甘草或光果甘草的干燥根和根茎的炮制加工品[1],为《中华人民共和国卫生部药品标准·中药成方制剂》第5 册收载的女宝胶囊的成分之一[2]。《伤寒杂病论》的260 个方剂中共有125 方涉及甘草[3],而在《伤寒论》中应用炙甘草者多达68 首[4]。所用炙甘草并非蜜炙甘草而是炒甘草,其本质是将甘草部分炭化[5‑6],且已经在荆芥炭、麦芽炭、枳实炭、蒲黄炭等炒炭的中药中发现了纳米类成分。这些纳米类成分具有较好的止血、降糖、镇痛、保肝、抗炎等功效。在甘草炭中也发现了纳米类成分,通过小鼠急性酒精性胃溃疡模型证明纳米类成分具有抗溃疡作用[7]。
目前,国家药品标准及各省市地方标准尚未收载甘草炭的炮制工艺及质量标准,药品生产企业使用的甘草炭均为厂家自行炮制,没有统一的炮制规范来控制其质量。本研究在传统炮制法的基础上,筛选出具有代表性、可行性的炮制方法,通过L9(34)正交试验进一步优化炮制工艺,参照《中华人民共和国药典》(以下简称《中国药典》)2020 年版(四部)相关方法,建立了甘草炭的炮制工艺及质量标准,为甘草炭饮片的质量控制提供参考。
1 材料
1.1 仪器
1260 型高效液相色谱仪(美国安捷伦公司);KQ‑300型超声清洗机(昆山市超声仪器有限公司);BT125D型电子天平(德国赛多利斯公司);MS‑5型炒药机(常州迈斯机械有限公司);CYJ900 型炒药机(山东创美机械科技有限公司);迷你型红外测温仪(深圳华盛昌机械实业有限公司)。
1.2 试药
对照品甘草苷(批号:111610‑201607,纯度:93.1%)、甘草酸铵(批号:110731‑201720,纯度:97.7%)均购自中国食品药品检定研究院;乙腈、甲醇均为色谱纯;水为自制超纯水。
甘草饮片为吉林省北药药材加工有限公司提供,批号为20180501,经四平市食品药品检验所副主任药师梁帅检定为正品,按照《中国药典》2020 年版(一部)甘草(甘草片)项下检验,各项均符合规定。
2 方法与结果
2.1 炮制工艺研究
2.1.1 影响因素及水平的选择 通过查阅文献[8‑11]及参考前期的研究工作,确定甘草炭的炒炭温度(红外测温仪测得炒药机内壁温度,A)、炒炭时间(B)和翻炒频率(C)作为考察因素。以甘草炭的水溶性浸出物、有效成分甘草苷和甘草酸的含量作为评价指标。采用L9(34)正交设计对甘草炭的炮制工艺进行优选,通过加权法进行评分,对试验结果进行分析。最后结合正交试验结果和炮制过程的评价筛选出既能满足甘草炭炮制目的,也符合现代药学理论的炮制工艺。
2.1.2 正交试验因素、水平的设计 根据前期的实验结果,甘草片质坚实、切面有一定的纤维性、部分有裂隙,炒炭温度在300 ℃以下长时间炒炭内部会先于外部炭化,需要提高温度短时间内迅速炒炭。选取大小均匀甘草片9 份,每份4 kg,置于炒药机内,按正交设计方案进行试验,得到各甘草炭炮制品,并采用综合评分法进行加权评分。炒炭要求存性,性状为判断炒炭样品是否符合要求的先决条件,在此基础上再进一步通过水溶性浸出物与指标性成分(甘草苷和甘草酸)的含量测定结果来细化炒炭条件。规定性状、浸出物和含量测定的权重系数分别为0.50、0.25、0.25,按公式(1)计算综合评分(Y)。
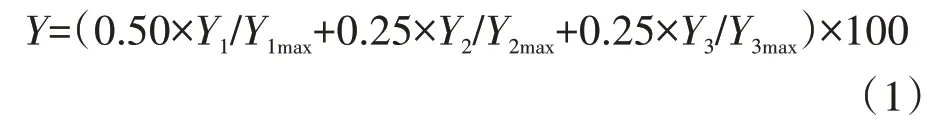
式中,Y1为性状赋分结果,Y2为浸出物结果,Y3为含量测定结果,Ymax为结果中最大值。
依据评分标准对不同条件下制备的炮制品进行打分,甘草炭性状按照外表面焦黑褐色、断面焦褐色为标准进行打分,浸出物和含量测定按照结果数值即分值进行打分,见表1~3。

表1 甘草炭炮制正交设计因素水平
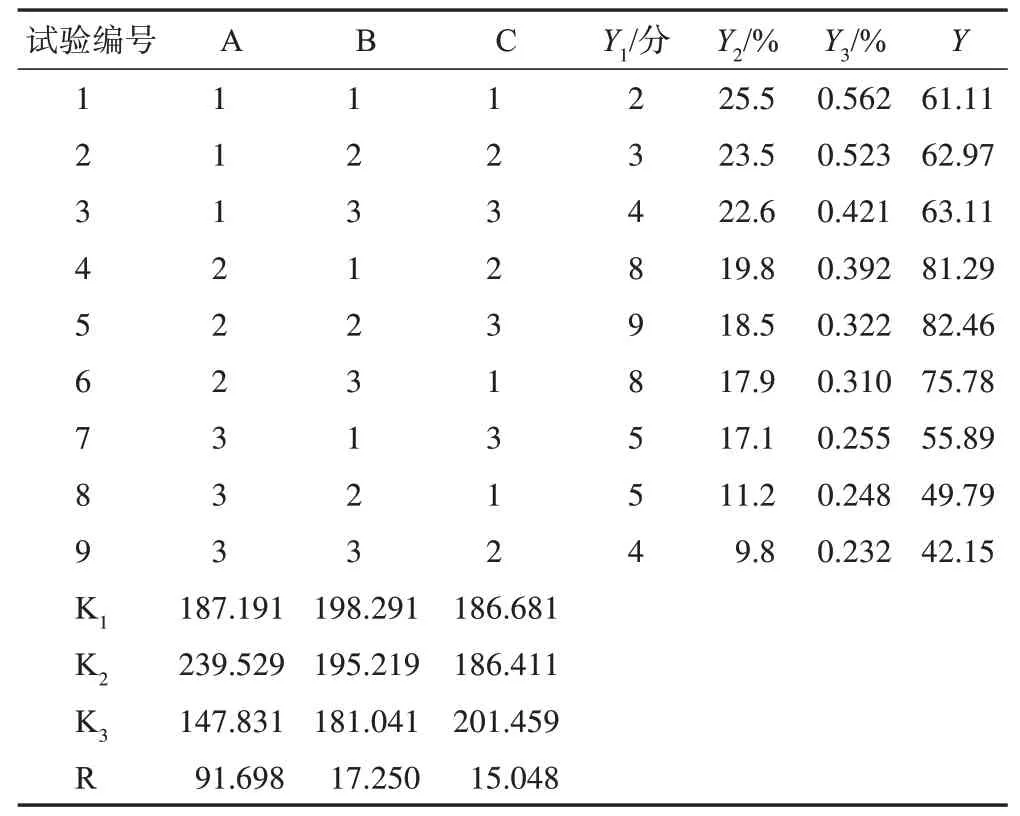
表2 甘草炭L9(34)正交试验因素水平分析

表3 甘草炭炮制结果方差分析
根据正交试验分析表和方差分析表可以得到,A 对实验结果的影响差异有统计学意义(P<0.05),而B、C对实验结果影响差异无统计学意义。翻动频率选择更快的C3,保证炒炭的样品的均匀性。综合评分结果,确定最佳工艺为A2B1C3,即温度为360 ℃,炒炭6 min,翻动频率25 r·min-1。
2.1.3 最佳炮制工艺条件的验证实验 选取甘草片4 kg,平行称取5份,按最佳炮制工艺制备甘草炭样品,计算浸出物和含量测定的值。结果见表4。

表4 最佳炮制工艺条件甘草炭验证实验结果 %
2.2 甘草炭质量标准
2.2.1 性状 本品呈类圆形或椭圆形的厚片,直径为0.6~3.5 cm,表面焦黑褐色,折断面呈焦褐色,略具焦香气,味微苦。
2.2.2 显微鉴别 粉末深棕色,纤维成束,直径为8~14 μm,壁厚,微木化,周围薄壁细胞含草酸钙方晶,形成晶纤维。草酸钙方晶多见,具缘纹孔导管较大。
2.2.3 薄层色谱 由于甘草炮制后成分含量发生变化,《中国药典》2020 年版(一部)甘草项下薄层色谱方法鉴别甘草及甘草酸单铵盐的斑点不明显,背景干扰较大,改为对甘草次酸指标的鉴别。取甘草炭粉末2 g,加盐酸2 mL、三氯甲烷15 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。另取甘草对照药材0.5 g,同法制成对照药材溶液。再取甘草次酸对照品,加乙醇制成含甘草次酸对照品2 mg·mL-1的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述3种溶液各1 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60 ℃)‑甲苯‑乙酸乙酯‑冰乙酸(10.0∶20.0∶7.0∶0.5)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙醇溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上显相同颜色的斑点(图1)。

图1 甘草炭薄层色谱图
2.2.4 检查总灰分 取10 批甘草炭供试品,照《中国药典》2020 年版(四部)通则2302 方法进行测定。10 批甘草炭的总灰分均在5.2%以下,较甘草片提高了约0.7%,故规定本品的总灰分不得超过6.0%(表5)。
2.2.5 浸出物 女宝胶囊中甘草炭为水提工艺,选择水作为提取溶剂,取10 批甘草炭供试品,照水溶性浸出物测定法[《中国药典》2020 年版(四部)通则2201]项下的热浸法测定,用水作溶剂进行试验。10 批甘草炭水溶性浸出物最高值为20.5%,最低值为17.1%,平均值为18.5%,拟定干品浸出物限度为不得少于14.0%(表5)。
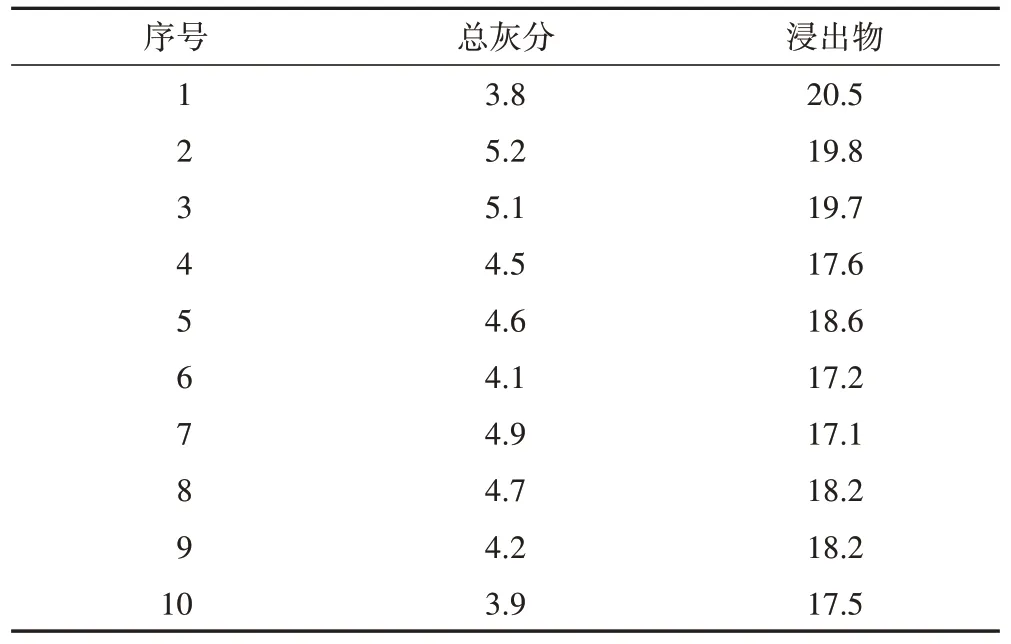
表5 甘草炭总灰分、浸出物测定结果 %
2.2.6 甘草苷和甘草酸含量测定
2.2.6.1 色谱条件 甘草炒炭后成分发生变化,调整梯度洗脱的溶剂比例以便分离。色谱柱:Sepax BR‑C18(250 mm×4.6 mm,5 μm);以乙腈为流动相A,0.05%磷酸溶液为流动相B,梯度洗脱(0~8 min,19%A;8~50 min,19%~42%A;50~55 min,42%~100%A;55~60 min,100%~19%A);流速:1.0 mL·min-1;波长:237 nm;柱温:30 ℃;进样量:10 μL。色谱图见图2。
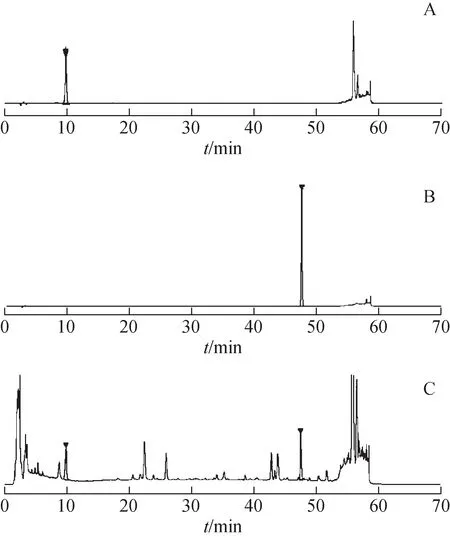
图2 对照品和甘草炭HPLC图
2.2.6.2 供试品溶液的制备 经考察,炮制后的甘草苷和甘草酸的含量明显降低,为使含量结果在合理的线性范围内,增加甘草炭的取样量,取甘草炭粉末(过三号筛)约2 g,精密称定,置具塞锥形瓶中,精密加入70%乙醇100 mL,密塞,称定质量,超声处理(250 W,40 kHz)30 min,放冷,再称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.2.6.3 对照品溶液的制备 称取甘草苷对照品11.80 mg,精密称定,置10 mL 量瓶中,称取甘草酸铵对照品(甘草酸质量=甘草酸铵质量/1.020 7)20.82 mg,精密称定,置20 mL 量瓶中,加70%乙醇使溶解并稀释至刻度,摇匀,即得。
2.2.6.4 线性关系考察 分别精密量取甘草苷对照品溶液0.2、0.4、0.6、1.0、1.5、2.0 mL置50 mL量瓶中,分别精密量取甘草酸铵对照品储备液0.25、0.50、1.00、2.00、4.00、6.00 mL 置10 mL量瓶中,加70%乙醇稀释至刻度,摇匀。分别精密吸取上述不同质量浓度对照品溶液各10 μL,注入液相色谱仪。以对照品峰面积为纵坐标(Y),对照品溶液质量浓度为横坐标(X)绘制标准曲线,计算回归方程,甘草苷:Y=15.357X+2.231 7(r=0.999 8),在4.39~43.94 μg·mL-1与峰面积线性关系良好;甘草酸铵:Y=5 784.7X-1.802 8(r=1.000 0),在25.43~610.20 μg·mL-1与峰面积线性关系良好。
2.2.6.5 精密度试验 取甘草炭(批号:20180501)供试品溶液,按2.2.6.1 项下方法连续进样6 次,测得甘草苷和甘草酸峰面积的RSD 分别为0.3%和0.3%,表明仪器精密度良好。
2.2.6.6 重复性试验 称取甘草炭(批号:20180501)供试品约2 g,精密称定,平行6 份,按2.2.6.2 项下方法制备,按2.2.6.1 项下方法测定,测得甘草苷和甘草酸含量的RSD 分别为1.0%和0.7%,表明方法重复性良好。
2.2.6.7 稳定性试验 取甘草炭(批号:20180501)供试品溶液,分别在0、2、4、8、12、18、24 h 按2.2.6.1 项下方法测定,考察供试品溶液稳定性,测得甘草苷和甘草酸含量的RSD 分别为1.0%和0.5%,表明供试品溶液在24 h内稳定性良好。
2.2.6.8 加样回收率试验 分别精密称定甘草苷对照品和甘草酸铵对照品16.05、40.94 mg,置1000 mL量瓶中,加70%乙醇使溶解并稀释至刻度,摇匀,作为甘草苷和甘草酸铵混合对照品储备液。称取甘草炭(批号:20180501)供试品(甘草苷质量分数为0.076%,甘草酸质量分数为0.206%)约2 g,精密称定,平行6 份,分别精密加入甘草苷和甘草酸铵混合对照品储备液100 mL,按照供试品溶液的制备方法制备,计算加样回收率,结果见表6,表明本方法回收率良好。
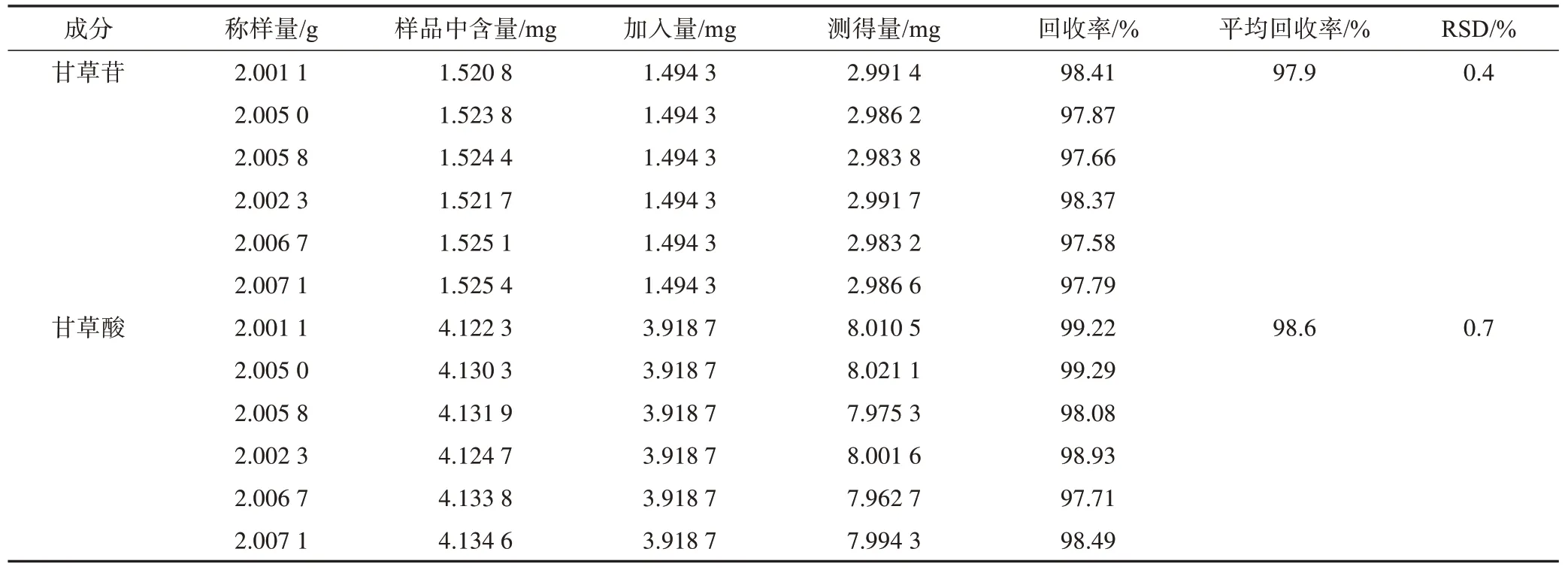
表6 甘草苷和甘草酸加样回收率试验结果
2.2.6.9 甘草苷和甘草酸含量 称取10 批甘草炭供试品,每批2 g,精密称定,按2.2.6.2 项下方法制备,按2.2.6.1 项下方法测定,计算甘草炭中甘草苷和甘草酸的含量,分析炮制前后的变化[12‑14]。结果见表7。
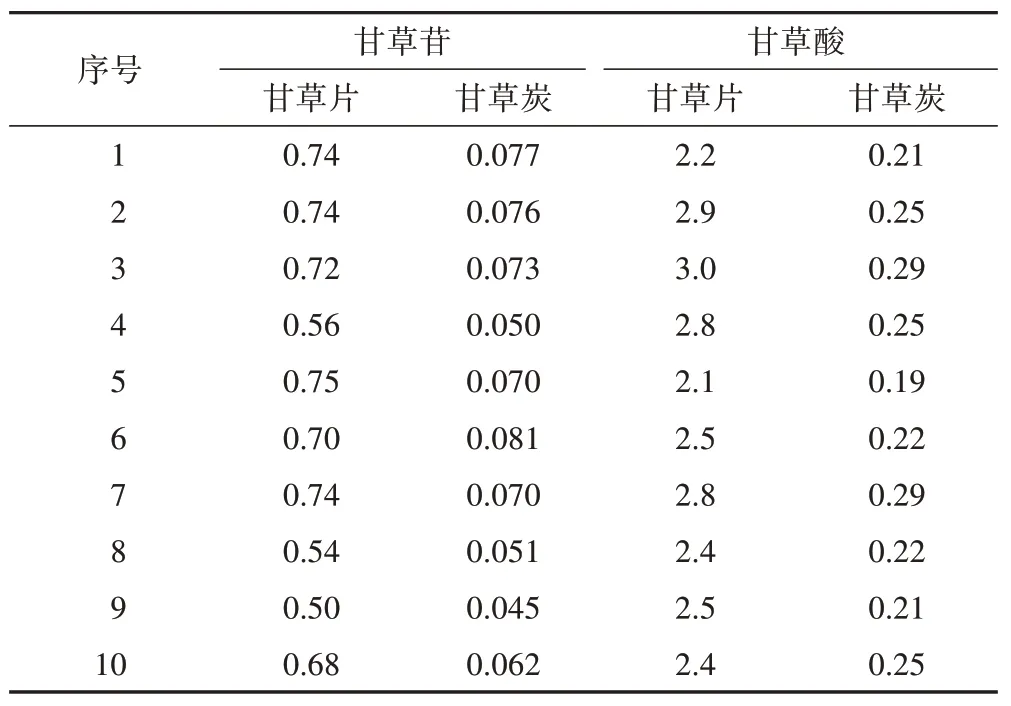
表7 甘草苷、甘草酸含量测定结果 %
10批甘草炭中甘草苷和甘草酸含量较炮制前明显降低,由甘草炭和甘草片的含量比值可知,甘草苷为炒炭前的9.8%,甘草酸为炒炭前的9.3%。根据《中国药典》2020年版(一部)甘草片中含甘草苷不得少于0.45%、甘草酸不得少于1.8%的限值,拟定甘草炭中甘草苷不得少于0.044%,甘草酸不得少于0.16%。
3 讨论
现代饮片加工生产对炮制工艺的各项指标提出了量化的要求,一些生产企业使用150~220 ℃的武火炒制的甘草炭无法达到炒炭的要求,传统中药炮制未对武火温度明确要求,应根据具体的品种调整火力,使饮片符合要求,达到炒炭存性的目的。
甘草炒炭后原有的成分及含量均发生较大变化,甘草苷和甘草酸的含量明显降低,在满足性状要求的同时,选择合理的含量作为甘草炭的评测指标,控制甘草炭饮片的质量。
