汉族与藏族健康人群肠道菌群结构的差异
2022-04-08毛明慧牛玥陈春李树春
毛明慧 牛玥 陈春 李树春
1中央民族大学药学院,民族医药教育部重点实验室(北京100081);2海西州天峻县织合玛乡卫生院(青海天峻817200)
人类的消化道内居住着许多微生物,胃肠道拥有最多数量和最多种类的微生物[1],总数估计在1013~1014[2]。肠道微生物群组成和功能的改变可以改变肠道通透性、消化和代谢以及免疫反应[3]。肠道菌群失衡可能导致许多疾病的发生,包括胃肠道疾病[4]、代谢性疾病[5]以及免疫[6]和精神疾病[7]。因此,肠道菌群在维持人类健康方面发挥着重要的作用。
由于遗传背景、饮食、文化习惯和社会经济等多种因素,人类肠道微生物群差异很大,不同种族或地域的人群肠道菌群构成存在差异[8]。其中,种族因素与多种微生物类群、基因家族和代谢途径密切相关[9],目前已有多项研究证明不同种族的人群肠道菌群差异较大。DESCHASAUX 等[10]人对生活在同一城市的不同种族人群进行16S rDNA,结果表明地理位置相同的不同种族的人群中肠道菌群组成存在显著差异。在另一项研究[2]中发现,汉族、藏族和回族学龄儿童肠道微生物群的α多样性具有显著差异。LIN 等[11]将汉族与非汉族群体进行分析比较,发现两个群体具有不同的菌群生物标志物和肠型。
中国是一个多民族的国家,诸多族群在历史的长河中相互融合、共同发展,形成了现有的56 个民族[12]。一般来说,每个民族都居住在不同的地理位置,并保留着各民族独特的饮食习惯和生活方式[13]。藏族生活在具有世界屋脊之称的青藏高原[14],由于地理上的分离,该地区的居民形成了与汉族人群不同的遗传背景,以及不同的文化和饮食习惯。因此,藏族和汉族人群适合于研究种族因素对肠道菌群的影响。此外,在现有研究中,有关其他民族与汉族之间的肠道菌群差异的研究较多,而藏族和汉族的肠道菌群差异研究较少。因此,基于种族、微生物群和健康差异之间的相互关系,本论文通过高通量测序和生物信息学分析探讨藏族和汉族肠道菌群差异,为从民族角度分析肠道菌群和疾病关系研究提供新的研究思路和实验支持。
1 资料与方法
1.1 样本和信息采集 本研究的样本采集于青海省天峻县健康志愿者。样本采集后30 min 内用粪便储存液保存,干冰运输到实验室后,置于-80 ℃冰箱中冷冻保存。纳入标准:汉族志愿者三代直系血亲均为汉族人群,藏族志愿者三代直系血亲均为藏族人群,无胃肠疾病、肝病、高血压或糖尿病等,近1 个月均未服用任何抗生素或微生物调节剂,遵循其传统生活方式和饮食习惯。最终纳入14 例汉族样本,28 例藏族样本。每个志愿者均签署知情同意书。本研究通过了中央民族大学伦理委员会的批准(批准号:ECMUC2019003CO)。
1.2 DNA 的提取和PCR 扩增 粪便肠道细菌DNA 的提取按照细菌基因组DNA 提取试剂盒MN NucleoSpin 96 Soi 使用说明进行操作。通过两步建库法,以DNA 为模板,设计带接头的引物进行PCR,利用PCR 产物为模板再次进行PCR。PCR体系(20 μL)为:纯化产物5 μL,MPPI-a 2.5 μL,MPPI-b 2.5 μL,2×Q5 HF MM 10 μL。反应条件为:90 ℃30 s,98 ℃10 s,65 ℃30 s,72 ℃30 s,72 ℃5 min,10个循环。将PCR 产物根据电泳定量结果,按质量比1∶1 进行混样。混样后,采用OMEGA DNA 纯化柱进行过柱纯化。1.8%的琼脂糖凝胶,经过电压120 V 的电泳40 min 后,切胶回收。最后使用Hiseq2500 上机测序。
1.3 生物信息学分析 测序得到的原始图像数据转化为序列数据,存储为fastq 文件格式。使用QIIME 软件对序列数据进行过滤,得到Clean tags,并构建稀释性曲线。使用USEARCH 得到用于后续分析的Final tags,并生成OUT丰度表使用CD-HIT软 件(http:∕∕www.bioinformatics.org∕cd-hit∕,Version 4.6.6)去除冗余,相似性阈值设置为90%,覆盖度阈值设置为90%,获得非冗余基因集。生成Alpha多样性表,使用R 语言ggplot2 包做盒型图。通过R 语言的vegan 包基于unifrac 距离进行β 多样性分析,进行组间Adonis 分析,检验组间β 多样性差异。通过R 语言的DESeq2 包和edge 包进行组间物种差异分析,利用韦恩图统计汉族和藏族差异OTU 的交并集情况。使用RDP 注释方法对OTU 进行物种注释。使用KRONA 对两组物种注释结果进行可视化展示。使用PICRUSt 进行LEfSe 分析并预测代谢途径,将OTU 表与greengenes 数据库对照,与KEGG 数据库进行比对,获得代谢通路信息。使用STAMP 软件进行组间代谢通路差异分析,得到两组间差异有统计学意义(P<0.05)的代谢通路。
1.4 统计学方法 研究样本数据均以均值±标准差表示,使用SPSS 软件的一般线性模型进行多变量分析。使用R 语言vegan 包进行db-RDA 环境因子分析。进行Adonis 分析,分析不同分组因素对样品差异的解释度。通过ANOSIM 分析,判断分组是否有意义。进行MRPP 分析,判断组间数据差异是否具有统计学意义。用R 语言vegan 包进行微生物环境因子分析,limma 包进行组间差异分析。P<0.05为差异有统计学意义。
2 结果
2.1 研究对象临床特征 两民族之间年龄和体质量指数(BMI)差异均无统计学意义(P>0.05),说明两组人群基本特征近似一致(表1)。
表1 藏族和汉族人群基本特征Tab.1 Basic characteristics of Tibetan and Han nationality±s
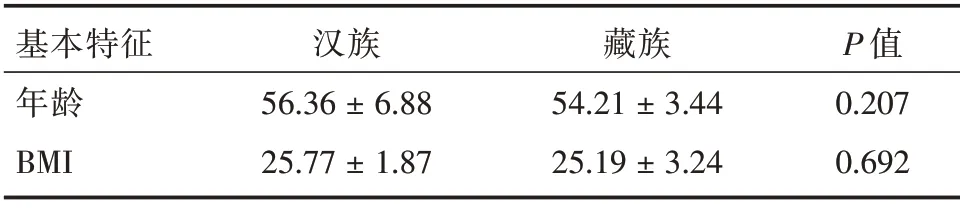
表1 藏族和汉族人群基本特征Tab.1 Basic characteristics of Tibetan and Han nationality±s
基本特征年龄BMI汉族56.36±6.88 25.77±1.87藏族54.21±3.44 25.19±3.24 P 值0.207 0.692
2.2 汉族和藏族肠道菌群的物种多样性 14 例汉族样本每个样本平均有51 505 条Final tags;28 例藏族样本每个样本平均有148 706 条Final tags。测序质量在QIIME 软件中生成Rank-Abundance 趋向水平直线,表明测序深度足够,测序数据可用于后续分析。α 多样性分析是对单个样本中物种多样性的分析,用来评价单个样本中物种的多寡(即丰富度)和各个物种的相对密度(即均匀度)。结果显示反映样品中群落丰富度的Shannon 指数和反映群落均匀度的Simpson 指数在两组间无显著差异[P(Shannon)=0.97,P(Simpson)=0.30](图1)。β 多样性分析结果显示汉族和藏族肠道菌群之间差异有统计学意义(P= 0.003,图2),两组样本菌群间的分化程度较高。
2.3 汉族和藏族肠道菌群差异 汉族和藏族共有OTU 497 个,汉族特有OTU 167 个,藏族特有OTU 287个(图3)。在筛选出的199个差异OTU中,在藏族显著上调的有164个,显著下调的有35个(图4)。注释到属水平的差异OTUs 中藏族优势菌属有瘤胃球菌属(Ruminococcus)、厌氧支原体属(Anaeroplasma)、变形杆菌属(Proteobacteria);汉族优势菌属有丁酸蓖麻单胞菌(Butyricimonas)、粪厌氧棒杆菌(Anaerostipes)、小杆菌属(Dialister)、巨球型菌属(Megasphaera)、巨单胞菌属(Megamonas)、考拉杆菌属(Phascolarctobacterium)、梭状杆菌(Clostridium)、链球菌(Streptococcus)、双歧杆菌(Bifidobacterium)、萨特氏菌属(Sutterella)、阿克曼氏菌(Akkermansia)、vadinCA02(图5)。

图3 差异OTU 分布韦恩图Fig.3 Venn diagram of differential OTU distribution

图4 火山图Fig.4 Volcano plot

图5 物种注释及菌群分布Fig.5 Species annotation and flora distribution
2.4 汉族和藏族人群生物标志物的鉴定 通过贝叶斯网络分析显示与民族关系最密切的菌群有6 个,属水平上有5个,分别是OTU82(Megamonas)、OTU80(Phascolarctobacterium)、OTU74(Megasphaera);种水平上有3 个,分别是OTU81(copri)、OTU56(stercorea)、OTU167(parainfluenzae)。其中与民族因素正相关的有普氏菌(copri)、粪普雷沃氏菌(stercorea),呈负相关的有副流感嗜血杆菌、巨单胞菌属(Megamonas)、考拉杆菌属(Phascolarctobac⁃terium)、巨球型菌属(Megasphaera)(图6A)。通过LEfSe 分析筛选出两组在属水平上菌群标志物为假丁酸弧菌(Pseudobutyrivibrio)、琥珀酸弧菌属(Succinivibrio)、伯吉古菌(Catenibaterium)、小杆菌属(Dialister)、血尿杆菌(Turicibacter)(图6B)。

图6 网络分析及生物标志物的鉴定Fig.6 Network analysis and identification of biomarkers
2.5 汉族和藏族人群差异代谢通路 两组的差异代谢通路共有10 个,其中,在汉族组中显著上调的通路有糖代谢(carbohydrate metabolism)、氨基酸代谢(amino acid metabolism)、脂质代谢(lipid metabolism)、新陈代谢(metabolism)、外源生物降解与代谢(xenobiotics biodegradation and metabolism)。在藏族组中显著上调的通路有酶家族(enzyme families)、复制和修复(replication and repair)、转化(translation)、核苷酸代谢(nucleotide metabolism)、萜类化合物和聚酮类化合物的代谢(metabolism of terpenoids and polyketides)(图7)。
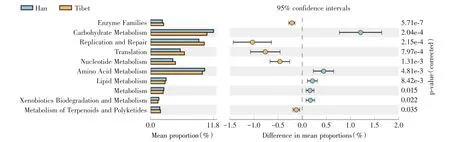
图7 功能预测分析Fig.7 Functional prediction analysis
3 讨论
目前,对影响肠道微生物群的主导因素还不清楚,但已知肠道微生物的形成涉及环境、遗传、种族和生活方式等因素[15]。已有研究[13]表明,种族因素对肠道菌群的影响显著。许多常见疾病也与肠道微生物的组成和种族有关[16-17]。中国的藏族和汉族在地理,饮食和遗传多样性有很大不同,这种差异会影响整个微生物群的组成。为了证实种族-微生物群假说,基于高通量测序数据,从物种和代谢通路角度较为全面的分析了藏族人群与汉族人群肠道菌群的差异,找到了属水平上两组的差异菌群,并通过贝叶斯网络筛选出与民族因素关联最密切的菌群。此外,本研究进一步筛选出了两组间具有显著差异的生物标志物和代谢通路。
从总体的物种丰度上看,藏族和汉族在门水平上的优势物种相同,均由拟杆菌门(Bacteroidetes),厚壁菌门(Firmicutes),放线菌门(Actinbacteria),变形菌门(Proteobcteria)组成,这和文献报道的一致[8]。其中汉族组的变形菌门丰度明显高于藏族。变形菌门中多数细菌对人体具有致病性,具有这种肠型的人更容易遭受外界病原体的感染[18]。SHIN 等[19]报道了变形杆菌门丰度与人类代谢紊乱、胃肠紊乱、炎症等的发生发展密切相关,可作为肠道失衡的标记物。
在属水平上,汉族组拟杆菌属(Bacteroides),毛螺菌属(Lachnospira),丁酸蓖麻单胞菌(Butyr⁃icimonas)、粪厌氧棒杆菌(Anaerostipes)、小杆菌属(Dialister)、巨球型菌属(Megasphaera)、巨单胞菌属(Megamonas)、考拉杆菌属(Phascolarctobacteri⁃um)、梭状杆菌(Clostridium)、链球菌(Streptococ⁃cus)、双歧杆菌(Bifidobacterium)、萨特氏菌属(Sut⁃terella)、阿克曼氏菌(Akkermansia)、vadinCA02 丰度明显高于藏族,而副杆菌属(Parabacteroides),罗氏菌属(Roseburia),多尔氏菌属(Dorea),颤螺菌属(Oscillospira),瘤胃球菌属(Ruminococcus)、厌氧支原体属(Anaeroplasma)、变形杆菌属(Proteobacte⁃ria)丰度明显低于藏族。其中,罗氏菌属是公认的有益菌[20],为丁酸产生菌[21]。多尔氏菌属也是产生丁酸的主要菌[22]。丁酸是短链脂肪酸的一种,可通过上调肠上皮紧密连接蛋白的表达,减少脂多糖、三甲胺等有害微生物代谢产物通过血肠屏障,对维持肠黏膜的完整性非常重要[23]。藏族的罗氏菌属和多尔氏菌属丰度显著高于汉族,说明藏族肠道菌群富含丁酸产生菌,这可能与藏族人群居住环境有关。拟杆菌属可以富集厌氧发酵类群,能够降解难降解底物,并将复杂多糖转化为单糖以生产ATP[24]。拟杆菌属丰度的变化可能与汉藏两组不同的饮食习惯有关,汉族人群多以碳水化合物为主,而藏族人群饮食习惯以肉类为主,果蔬摄入量较少,脂肪及蛋白质摄入量高于汉族人群[25]。
有文献[24]报道发现肠道富含阿克曼氏菌的患者具有更好的免疫治疗效果,阿克曼氏菌也有助于抑制肥胖和改善酒精相关性肝硬化。熊菲等[26]研究发现双歧杆菌通过胃肠道途径可能促进树突状细胞的分化、发育、成熟,可能增强树突状细胞的肠道免疫作用;在树突状细胞分化发育过程中发挥的作用可能也是双歧杆菌影响机体免疫功能的重要环节。这表明汉族和藏族人群的肠道菌群差异可能导致汉族和藏族人群某些疾病的发病率不同。
本研究通过LEfSe 分析最终筛选出两组在属水平上菌群标志物为假丁酸弧菌(Pseudobutyri⁃vibrio)、琥珀酸弧菌属(Succinivibrio)、伯吉古菌(Catenibaterium)、小杆菌属(Dialister)、血尿杆菌(Turicibacter)。在功能和代谢通路分析中,我们发现两组共有十条差异代谢通路。结果表明,糖代谢、氨基酸代谢、脂质代谢、新陈代谢等代谢通路在藏族组显著下调。这可能与藏族人群常年生活在高海拔寒冷地区,高脂饮食,新陈代谢慢有关[27]。
综上所述,本研究结果表明汉族和藏族人群肠道菌群的种属、基因功能和代谢通路存在显著差异,意味着在将微生物群差异与疾病联系的研究中,需要考虑种族差异作为潜在的混杂因素。然而,种族可能是一个混合因素,不同种族间的遗传、环境和饮食差异都对肠道微生物群的组成有一定影响。此外,不同因素之间存在共同影响。因此,这些结果的进一步研究方向,可能是探索肠道微生物群变化的内外因素,并建立各种潜在因素的相互作用网络;发现更多微生物族群的生物标记物,从而将其作为健康差异的潜在诊断或治疗方法。
本研究还存在许多不足之处,由于临床所限,纳入的样本量较少,两组研究对象的男女比例有差异,研究纳入的均为年龄大的受试者,缺少年轻受试者。由于16S rDNA 技术的限制,本研究检测范围不如宏基因组测序范围广。在未来的研究中,将使用更科学的测序技术,在扩大样本量的基础上,去除混杂因素后进一步研究民族差异对肠道菌群的影响机制。
