脑血管病基因组学数据分析流程建设
2022-04-06许喆程丝刘阳石延枫李昊
许喆,程丝,刘阳,石延枫,李昊
脑血管病是我国第一大死亡原因[1],且具有病因分型多[2]、遗传架构(genetic architecture)复杂等特点[3]。随着高通量测序和质谱技术的进步,在脑血管病分子机制研究中,基因组、转录组、表观遗传组、蛋白质组、代谢组、宏基因组等组学技术被应用[4-6],融合各组学技术的多组学分析已经成为未来医学和生命科学研究的趋势[7-8]。在众多组学提供的不同维度的数据中,基因组信息在体内最为稳定,组织特异性低,因此成为多组学研究重要的切入点[7],应用也最为广泛[9-15]。但是由于研究目标、样本选择有所不同,脑血管病领域各个基因组学研究的分析方案不尽相同,导致脑血管病领域基因组数据生物信息学分析流程整合度不高,缺乏系统、全面的汇总。
本研究在临床需求和文献调研的基础上,梳理了脑血管病基因组学研究中常用、主流、稳定的分析方案,以模块化的设计思路,以中国国家卒中登记Ⅲ(China national stroke registry-Ⅲ,CNSR-Ⅲ)研究的基因组学数据为测试集[16-17],搭建标准化的生物信息学分析流程,为脑血管病临床和基础研究提供大数据支撑。
1 方法
1.1 确定分析流程所需模块和技术参数
1.1.1 文献复习 通过查阅MEGASTROK E(multi-ancestry genome-wide association study of stroke)项目、GENS(GENetics of Stroke)登记研究、CHARGE(cohorts for heart and aging research in genomic epidemiology)合作组研究的相关文献[15,18-19],总结脑血管病队列和群体遗传学研究中常用的分析策略、研究方案设计原则、数据质控方案和管理模式等,收集生物信息学分析的技术参数。
1.1.2 专家咨询 咨询神经病学、神经生物学、流行病学专家,调研临床诊断和基因检测对基因组学、生物信息学数据分析的需求。向生物信息学、群体遗传学、基因组学、生物样本库等领域专家,咨询基因组学数据分析中合理的功能模块和实现方案。
1.1.3 数据库和在线工具调研 调研生物信息学、基因组学、表观遗传学、转录组学等多组学数据库,下载相应数据或者优化检索方式,将数据库的内容或信息嵌入分析流程。调研数据库包括RegulomeDB[20]、GTEx(genotypetissue expression)[21]、GeneHancer(genomewide integration of enhancers and target genes in GeneCards)[22]、ClinVar(public archive of interpretations of clinically relevant variants)[23]、OMIM(online mendelian inheritance in man,https://omim.org)、Orphanet(www.orpha.net)等。同时调研了Galaxy等在线分析工具[24],考察基因组学数据标准化分析流程设计思路。
1.2 高性能运算集群 基于首都医科大学附属北京天坛医院高性能运算集群,开发和搭建生物信息学分析流程。集群包括80台1路、76台2路、4台4路、1台8路CPU计算节点,9台GPU计算节点,理论浮点运算能力375万亿次/秒。采用中科曙光parastor 200并行存储系统,提供5.3 PB可用存储空间。集群使用1套100 Gbps Infiniband网络,1套千兆管理网络,提供1台登录节点,提供用户登录、编译软件、提交作业、上传下载数据等功能;提供1台管理节点,安装Gridview作业管理系统,并提供集群监控功能。
1.3 流程测试和优化 基于CNSR-Ⅲ研究等队列的基因组学数据,对流程进行测试。测试样本为CNSR-Ⅲ研究队列遗传亚组共12 603例缺血性脑血管病患者外周血白细胞DNA[17]。这些DNA样本被用于进行全基因组测序,生成的遗传数据经过本研究搭建的生物信息学流程处理,保留测序数据质量合格、无污染、无亲缘关系、无显著遗传背景差异的样本,用于后续的群体遗传学、多组学数据挖掘。在测试过程中,基于高性能运算集群的架构和各个计算节点的算力,结合数据量、单个运算任务对内存需求、不同模块中生物信息软件的算法和并行计算的参数、集群带宽等,拆分计算任务,使集群算力得到高效应用。
1.4 数据管理方案调研 遵照《中华人民共和国生物安全法》《中华人民共和国人类遗传资源管理条例》及《首都医科大学附属北京天坛医院人类遗传资源管理办法》的相关规定,对研究涉及的数据和样本进行管理。与国内相关研究团队的数据管理部门、相关专家进行学术交流,学习人类遗传资源管理经验。
2 结果
本研究搭建的基因组学数据生物信息学分析流程,主要包括组学大数据质控和预处理、群体遗传学质控和样本清理、临床相关位点解析等部分,各部分又按照研究目的和功能、使用软件和软件组合的不同,细分为不同模块(图1)。不同模块之间可以自由组合,增强了本流程的兼容性和普适性。此外,本流程在搭建过程中也考虑到研究方案顶层设计和项目管理的需求,在项目论证和规划阶段引入基因组学、生物信息学专家,以便明确研究目标,选择合理的组学检测技术。
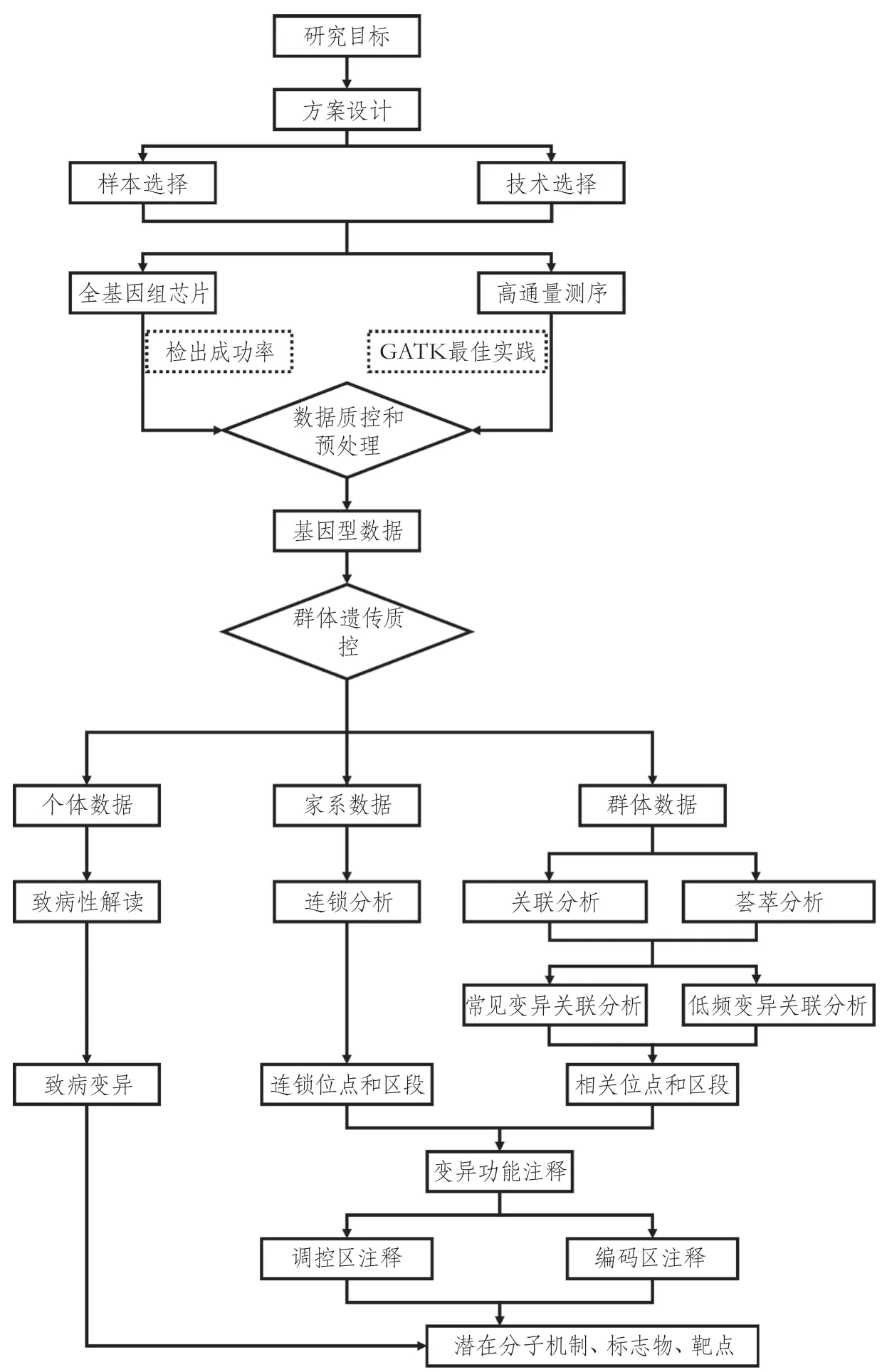
图1 基因组学数据分析流程
2.1 检测技术选择 生物信息分析人员根据医师科学家的研究目的,协商选取合适的检测技术,预先确认分析方案。检测技术的选择遵循以下原则:①优先选择数据质量可靠,稳定性、重复性好,认可程度高的检测技术;②选择样本消耗量适中的检测技术,兼顾数据和样本用于后续其他研究的可能;③对于个体化订制的检测方案(如探针和靶向捕获试剂盒等),需要进行预试验,评估检测体系运转情况和数据质量,利用预试验产生的基因组学数据,对分析流程进行测试和优化;④项目的检测方法一旦确定,不宜中途调整,检测试剂和仪器也应确保是同一型号、相同批次,避免检测结果出现批次效应。
2.2 原始数据预处理与质量评估
2.2.1 高通量测序数据 高通量测序作为目前获取样本最全面遗传信息的主流技术手段,被广泛应用于复杂疾病的基因组学分析、孟德尔遗传病的分子诊断等领域。通常情况下,高通量测序的实验由专业的检测机构或实验室完成,检测结果以测序原始数据(FASTQ格式)和检测报告的形式向临床研究人员反馈。
在测序原始数据复制、传输、备份的全部环节,都需要核对数据完整性,避免数据文件损坏。数据每复制一次,检测原文件和复制本的MD5码,两文件的MD5码必须完全一致。
而后,按照GATK(genome analysis toolkit)最佳实践的高通量测序数据检出种系变异中的单核苷酸变异(single nucleotide variants,SNV)和插入缺失多态(insertion-deletion,INDEL)的处理流程(图2)[25],对测序原始数据进行预处理,同时完成测序深度、覆盖情况、微生物污染(GC含量)、人源污染等方面的评估。
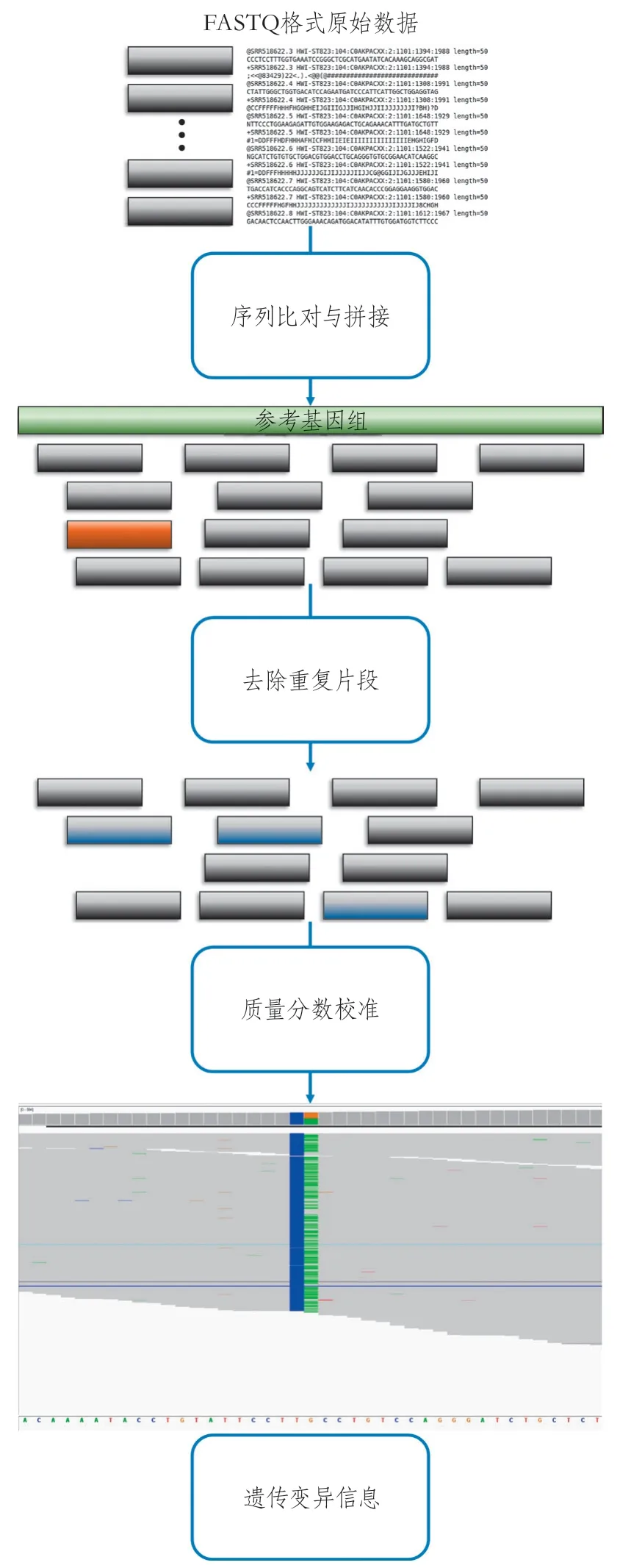
图2 GATK最佳实践流程
2.2.2 分型数据 分型是指获取样本预设变异位点基因型的操作,通常是指获取单核苷酸多态(single nucleotide polymorphism,SNP)位点的基因型。根据通量和分型原理的不同,采用不同的方案进行预处理。
对于通量较高的全基因组SNP分型芯片,使用芯片厂家指定的软件,如GenomeStudio(Illumina)和Genotyping Console(Affymetrix)进行SNP分型芯片的数据预处理。在生产厂商的网站找到相应型号芯片的解析文件,用于原始数据的导入和可视化。而后需评估各个样本的质量,如果某样本在全基因组上被成功分型的位点数目低于芯片全部预设位点数目的95%,则认为该样本DNA发生降解,故而将该样本剔除。在剩余样本组成的群体中,再进行位点水平的质量评估,如果一个位点有超过3%的样本没有分型成功,分析该位点的统计学效力不足,这些位点也需剔除。
对通量较低的候选SNP位点进行分型,如荧光探针分型(TaqMan)、质谱分型(Agena Bioscience)、KASPar®等[26-28],受上样量、检测仪器、操作熟练程度等外部因素影响较大,分型成功率的阈值可降低到80%。
2.2.3 污染评估 可控和不可控因素引入的样本污染,将降低来自污染样本基因组学数据的可信度,因此污染样本需要从研究群体中删除。
样本污染主要来自微生物和人源DNA。被微生物污染的样本,测序得到的基因组GC含量明显异于正常人类基因组GC含量(39%~45%),因此可以通过全基因组测序数据预处理、计算GC含量来判断。人源DNA污染会导致被检测样本的基因组上存在过多的杂合变异,可使用VerifyBamID软件判断全基因组测序和芯片分型样本人源污染情况[26]。2.3 群体遗传学质控 无污染、遗传变异检测成功率合格、数据量满足要求的样本,可以按照研究目标的不同,开展面向群体的关联分析、面向家系的连锁分析,以及面向单基因病个体的突变致病性注释等研究(图1)。其中用于关联分析、连锁分析的样本,需要进行群体遗传学质控。对于关联分析,研究群体中不得存在重复样本、具有亲缘关系的样本或遗传背景明显离群的样本,同时评估群体的亚分层等群体遗传结构;对于连锁分析,需通过群体遗传质控确定家系中没有样本遗传数据与亲缘关系不匹配的情况。使用RelPair、KING等软件[29-30],分析样本间的亲缘关系以及样本有无重复。对于关联分析,以保留尽可能多的样本进入分析流程为原则,去掉具有亲缘关系的样本对中的一个样本,确保纳入分析的样本中不存在具有3度以内的亲缘关系样本。
遗传背景的离群值和人群亚分层使用主成分分析进行。在全基因组上选取没有连锁不平衡关系的SNP位点,使用GCTA软件进行主成分分析[31],使用STRUCTURE软件评估人群遗传结构[32]。本分析流程使用千人基因组数据库中的中国南北方汉族,或欧洲人群、非洲人群作为参考人群[33],对目标群体的样本进行主成分分析和聚类分析。如果目标群体在前二或前三个主成分中有明显的离群个体,或者明显和其他族裔人群聚类接近,则将该样本删除。如果人群具有明显的分类,需要在后续的关联分析中,将该样本的前2~10个主成分作为协变量,以降低人群亚分层对关联分析结果的影响。
上述群体遗传学质控只能使用覆盖全基因组的SNP位点来进行,因此对于候选基因SNP分型的项目和样本,无法进行相应的群体遗传质控。
在CNSR-Ⅲ研究队列全基因组测序样本中,完成群体遗传学质控后,最终获得无3度以内亲缘关系、遗传背景离群和亚分层的研究人群,样本共计10 241例,这些样本可用于全基因组关联分析。
2.4 变异功能注释 注释是根据遗传变异在基因组上的物理位置、变异形式等信息,解析和预测该遗传变异的生物学功能的流程。根据注释方法和内容的不同,注释在蛋白编码和基因表达调控两个水平上展开。
2.4.1 蛋白编码区注释 位于基因的编码区遗传变异会直接改变密码子的组成,对蛋白编码造成影响。同时,基因内含子区的可变剪切位点发生变异,会影响蛋白编码中外显子和开放阅读框的选取,也会影响蛋白质中的氨基酸序列排布。选取VEP(ensembl variant effect predictor)和ANNOVAR(annotation of genetic variants)软件[34-35],对基因编码区的变异进行注释,使用dbscSNV数据库和SpliceAI软件对可变剪切位点的变异功能进行注释和预测[36-37]。
2.4.2 表达调控区注释 基因组上超过97%的序列为非编码区,但是这些序列可能执行调节基因表达的功能[38]。这些区域发生的DNA变异有可能改变转录调节因子的结合能力,从而影响基因表达。对于表达调控区变异的注释,主要通过检索RegulomeDB、GTEx、GeneHancer等数据库来完成[20-22]。通过判断遗传变异是否位于表达调控区预测对转录调节因子结合能力的影响,判断遗传变异对基因表达的潜在影响。
2.4.3 变异致病性评估 对于单基因病患者的基因组研究,主要目标是确定导致患者发病的变异。因此对该类患者携带的遗传变异,除了进行功能注释,还需要进行致病性评估,在实践中采用软件评估和数据库检索两种方案进行。软件预测采用InterVar等软件[39],按照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)标准[40],进行变异的致病性评估。数据库检索是从ClinVar[23]、OMIM(https://omim.org)、Orphanet(www.orpha.net)等数据库中检索患者携带的遗传变异是否为数据库已收录的致病变异,并分析患者表型与数据库收录的表型是否相符。
2.5 连锁分析 连锁分析是在疾病家系中,按遗传多态和疾病表型的共分离关系,定位疾病致病变异的分析(图1),该分析在具有亲缘关系的家系样本中展开。可以使用的软件包括Merlin(multipoint engine for rapid likelihood inference)、Haplo2Ped等[41-42]。需要注意的是,连锁分析通常是将遗传变异作为标注基因组区段的分子标记,进行致病基因所在区段的定位,并不能直接定位致病突变。因此,须对连锁分析得到的阳性区段内所有遗传变异进行注释和精细定位,并进行分子生物学、细胞生物学实验,对可能致病的遗传变异进行功能验证,才能确定致病变异。
2.6 关联分析 关联分析是指以不同个体的遗传变异信息作为自变量,个体的表型信息作为因变量,在群体层面研究遗传变异和表型的相关关系的方法[43]。按照遗传变异在群体中的频率不同,分别进行常见变异关联分析和低频变异关联分析。
2.6.1 常见变异关联分析 常见变异是指次要等位基因频率(minor allele frequency,MAF)在群体中>1%,或者在研究样本中携带者数量超过一定阈值(如30个)的遗传变异。常见变异关联分析采用广义线性模型对遗传数据和性状信息进行线性拟合,对于数量性状和质量性状,分别采用线性回归和logistic回归的方法。常见变异关联分析使用PLINK软件开展[44],以单独一个遗传变异位点为单位进行计算,估计其与表型的相关性。
在常见变异关联分析的过程中,由于遗传变异测序或分型失败,导致一部分样本的个别位点,或者样本群体的部分位点基因型数据缺失。这时可采用基因型填补(impute)方法,推测缺失位点的基因型。该操作基于参考群体的基因组遗传变异和数据缺失的遗传变异之间连锁不平衡关系,推断基因型已知的遗传变异位点周边基因组区域中,基因型未知或未被分型的遗传变异的基因型。该操作对于常见变异表现较好,但是能够被推定基因型的遗传变异数量有限。
在常见变异关联分析中,基因型填补能够增加基因组的覆盖范围和密度,提高用于关联分析的遗传变异数量。在本流程中,使用IMPUTE2软件及其相应的参考人群数据,完成基因型填补过程[45]。
2.6.2 低频变异关联分析 低频变异是指MAF<1%,或者在研究样本中携带者数量低于一定阈值(如30个)的遗传变异。尽管低频变异的数量众多,但从单个遗传变异的频率来看,携带者数量较少,因此不适宜采用常见变异关联分析中,以单个变异为单位进行关联分析的方法。而是将一段基因组区域内的全部低频变异进行归纳,依据频率等因素对每个低频变异进行加权,构建统计量,再与表型进行关联分析。在本流程中,使用SKAT、rvtests等软件[46-47],以基因组区段为单位,进行低频变异关联分析。
2.6.3 荟萃分析 整合不同研究的全基因组关联分析结果进行荟萃分析,可以提升样本量,增加统计学效力。对于可以开展荟萃分析的项目和数据,使用METAL和RARE METAL软件[48-49],进行常见变异荟萃分析和低频变异荟萃分析。
2.7 跨组学研究 关联分析得到的结论往往只能表明相关性而不是因果关系,因此关联分析的研究结果难以实现临床转化。在大数据、多组学时代,可以通过跨组学分析的方法,证明因果关系或者中介因素。使用遗传多态作为分组依据,模仿随机对照试验对样本分组进行的孟德尔随机研究,能够推断来自不同组学的表型数据之间的因果关系。为此,在流程中设计基于CIT包的因果推断检验[50],以及基于TwoSampleMR包的孟德尔随机分析[51]。
3 讨论
脑血管病是严重威胁我国国民健康的复杂疾病,对脑血管病分子机制的研究,有助于发现潜在的药物靶标,开发新的治疗方案。以高通量测序和质谱为代表的组学技术蓬勃发展,推动了脑血管病在基因组、转录组、表观遗传组、蛋白质组、代谢组、宏基因组等领域研究的长足进步,加深了对脑血管病分子机制的认识。但是由于种种限制,以往研究样本量较小、组学数据维度单一,通常只能证明相关性而非因果关系,因而对临床治疗的帮助有限。在大样本量队列中展开多组学分析,不仅能够从多个维度揭示疾病分子机制,也能更清楚地确定生物标志物、药物靶标和疾病结局的因果关系。多组学是融合了基因组、转录组、表观遗传组、蛋白质组、代谢组、宏基因组等多个组学的综合性学科。一次性开展多个组学的研究,成本巨大而且风险较高,因此在多组学研究中需要进行良好的顶层设计和分析,以期为脑血管病的临床治疗和研究提供新的支持[52]。
以往生物信息学标准化分析流程的研究中,对于基因组数据的标准化分析关注较少[53]。以往脑血管病的基因组学研究,由于目标、组学技术和数据的差异,分析策略和流程的选择较为单一。因此,基因组学数据的分析流程缺少有效的集成和优化,难以满足脑血管病等多种复杂疾病的研究需求[54]。不同亚型的脑血管病临床表现难以区分,单基因病和多因微效型脑血管病患者临床表现相似,增加了分型诊疗的难度。对于脑血管病的大规模测序研究,建议先对单基因病型脑血管病患者进行致病性评估和连锁分析,为临床诊断定位可能的致病突变。在关联分析中,优先保留多因微效型脑血管病患者的资料,增强样本遗传架构的一致性和研究的统计效力。为了分析CNSR-Ⅲ研究队列基因组学数据,在调研研究需求和脑血管病特点的基础上,搭建了基因组学数据分析流程。该流程以模块化设计思路,包含了关联分析、连锁分析、遗传变异功能注释、致病性评估、跨组学分析等多种分析方法,能够满足脑血管病研究所需的单基因病患者评估、关联分析等功能。可以根据不同的研究目的,有选择地使用本流程的不同模块。本流程为分析脑血管病基因组数据提供了全面、系统的方案。同时,本研究关注了以往研究论文较少提及的高通量组学数据的管理方案,介绍了组学数据接收、核对、安全管理的原则,有助于脑血管病领域基因组学和多组学研究的开展。
本研究建立的分析流程在以下方面需要继续优化:首先,本流程主要用于基因组DNA遗传变异的检出和研究,对多组学包含的其他组学数据,如转录组、表观遗传组、蛋白质组、代谢组等研究产出的数据,尚未搭建分析流程。其次,本流程适合于基因组DNA发生的种系变异,不适用于体细胞变异的分析。在CNSR-Ⅲ研究后续的基因组等多组学研究中,随着研究的深入和其他组学数据的产生,将逐步完善分析流程。最后,需要指出的是,尽管能够通过流程,提升数据分析效率,但是在临床科研的实际操作中,具体的参数设置,如协变量、病例-对照分组等,都会对分析结果产生影响。因此在分析过程中,仍然需要根据临床和生物学知识,调试相应参数以获取合理的分析结果。
说明:本文涉及的部分生物信息学术语或数据库名称在国内尚无统一译文,强行将这些术语或名称翻译成中文将影响读者对原意的理解,因此本文对此类术语及名称未进行翻译。
【点睛】标准化、模块化生物信息分析流程的确立,为全面挖掘脑血管病基因组学数据、展示其蕴含的致病信息提供了系统性解决方案,将推动脑血管病多组学研究的发展。
