高效合成L-高丝氨酸大肠杆菌基因工程菌株的构建
2022-03-30林蓓蓓张稳杰徐皓然张成林
张 宇,夏 利,林蓓蓓,张稳杰,徐皓然,张成林
(天津科技大学生物工程学院,天津 300457)
L-高丝氨酸属于L-天冬氨酸族非蛋白质氨基酸,是合成必需氨基酸L-苏氨酸、L-甲硫氨酸和L-异亮氨酸的前体物[1]。作为重要的平台化合物,L-高丝氨酸还被用于O-乙酰高丝氨酸、L-氮杂环丁烷、高丝氨酸内酯、异丁醇和γ-丁内酯等高附加值化合物的合成[2-7]。此外,有研究表明L-高丝氨酸还具有提高植物抗逆性、促进家禽生长的功能[5,8]。由此可见,L-高丝氨酸有望广泛应用于食品、医疗及农业等领域。
目前L-高丝氨酸主要通过化学法合成,但由于合成效率低等原因限制了其生产规模和应用范围[3]。近年来,随着代谢工程和合成生物学的迅猛发展,发酵法合成L-高丝氨酸受到广泛关注。如图1所示,L-高丝氨酸的生物合成以草酰乙酸为前体物,经天冬氨酸激酶、高丝氨酸脱氢酶和天冬氨酸半醛脱氢酶催化合成,其中天冬氨酸激酶和高丝氨酸脱氢酶为限速酶[4]。L-高丝氨酸通常在胞内不积累,而直接代谢为L-苏氨酸。此外,L-赖氨酸与L-高丝氨酸竞争前体物天冬氨酸半醛。故常见L-高丝氨酸生产菌株代谢工程选育策略主要包括增强前体物草酰乙酸合成、阻断L-高丝氨酸竞争和降解途径等。Plachý等[9]最先报道利用L-苏氨酸营养缺陷型谷氨酸棒状杆菌(Corynebacterium glutamicum)发酵72 h生成14.2 g/LL-高丝氨酸,但同时积累7.5 g/LL-赖氨酸。Li Ning等[4]利用阻断C. glutamicum L-高丝氨酸竞争及降解途径并增加其合成通量等策略,使得L-高丝氨酸产量达到8.8 g/L。但总体而言,利用C. glutamicum合成L-高丝氨酸效率较低。
大肠杆菌(Escherichia coli)因遗传背景清晰、生长速度快等特性也被应用于发酵法合成L-高丝氨酸研究[10-17]。Li Hua等[16]报道通过代谢工程改造E. coli合成L-高丝氨酸39.5 g/L(45 h),为目前报道的最高产量。Liu Peng等[17]通过对E. coli代谢网络重构使得L-高丝氨酸产量达37.6 g/L(108 h)。尽管利用E. coli合成L-高丝氨酸取得了显著进展,但构建适用于工业化生产的细胞工厂仍具有挑战性。主要表现在,为使L-高丝氨酸积累,现有菌株的L-高丝氨酸降解途径相关基因均被敲除,造成L-赖氨酸、L-苏氨酸、L-异亮氨酸、L-甲硫氨酸等多种营养物质缺陷。故需在发酵培养过程中添加适量的上述营养物质。这不仅增加了生产成本,还使得发酵过程复杂化。
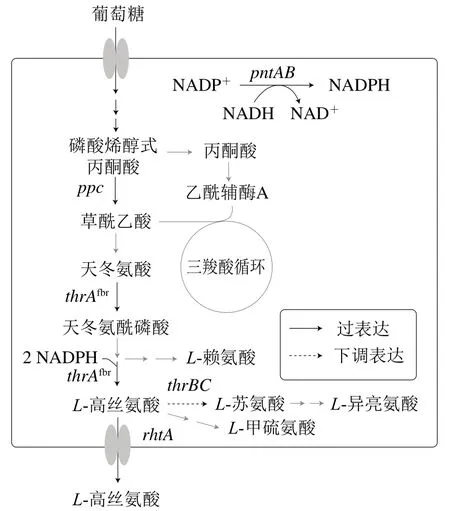
图1 L-高丝氨酸合成途径及本研究主要策略Fig. 1 Pathway and strategy for the synthesis of L-homoserine used in this study
本研究针对上述问题,以E. coliW3110为底盘细胞构建非营养缺陷型的L-高丝氨酸高效合成基因工程菌。首先弱化L-高丝氨酸降解途径削弱其降解;然后通过增强合成代谢流、提高前体物和辅酶供应实现L-高丝氨酸积累;在此基础上促进L-高丝氨酸外排,进一步提高其产量(图1)。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
本研究使用的菌株和质粒见表1。其中E. coliDH5α用于质粒构建,E. coliW3110 ΔlacI作为出发菌株用于构建L-高丝氨酸生产菌株。E. coliCRISPR/Cas9基因组编辑系统所用质粒pRedCas9和pGRB由天津大学陈涛教授惠赠。

表1 菌株和质粒Table 1 Strains and plasmids used in this study
1.1.2 培养基
LB(Luria-Bertan)培养基:蛋白胨10 g/L,酵母粉5 g/L,NaCl 10 g/L,pH 7.0~7.5。
斜面活化培养基:葡萄糖5 g/L,蛋白胨10 g/L,酵母粉5 g/L,牛肉膏10 g/L,NaCl 5 g/L,琼脂20 g/L,pH 7.0~7.5。
摇瓶种子培养基:葡萄糖20 g/L,蛋白胨5 g/L,酵母粉12 g/L,KH2PO41.2 g/L,MgSO4·H2O 0.5 g/L,FeSO4·7H2O 5 mg/L,MnSO4·7H2O 8 mg/L,VB12 mg/L,pH 7.0~7.5。发酵罐种子培养基同摇瓶种子培养基。
摇瓶发酵培养基:葡萄糖20 g/L,蛋白胨5 g/L,酵母粉4 g/L, KH2PO42 g/L,MgSO4·H2O 1 g/L,FeSO4·7H2O 10 mg/L,MnSO4·7H2O 10 mg/L,pH 7.0~7.5。
发酵罐发酵培养基:葡萄糖30 g/L,蛋白胨8 g/L,酵母粉5 g/L,KH2PO43.5 g/L,MgSO4·H2O 1 g/L,FeSO4·7H2O 10 mg/L,MnSO4·7H2O 10 mg/L,玉米浆30 mL/L,pH 7.0~7.5。
1.1.3 引物
引物由苏州金唯智生物科技有限公司合成,序列见表2。

表2 DNA片段扩增引物Table 2 Primers used for the amplification of DNA fragments

续表2
1.1.4 试剂
(NH4)2SO4、MgSO4·H2O、KH2PO4国药集团试剂有限公司;L-高丝氨酸 上海阿拉丁生化科技股份有限公司;胰蛋白胨、酵母提取物 英国Oxoid公司;基因组提取试剂盒、质粒提取试剂盒、胶回收试剂盒美国Omega BioTek公司;限制性内切酶、Primer STAR HS DNA聚合酶 宝日医生物技术(北京)有限公司;一步法克隆试剂盒 南京诺唯赞生物科技有限公司;甲醇、乙腈(均为色谱纯) 美国赛默飞世尔科技公司。
1.2 仪器与设备
U3000高效液相色谱仪 美国赛默飞世尔科技公司;PTC-1148型聚合酶链式反应(polymerase chain reaction,PCR)仪 美国Bio-Rad公司;ZORBAX Eclipse AAA氨基酸柱 美国Agilent公司;SBA-40D生物传感器分析仪 山东省科学院生物研究所。
1.3 方法
1.3.1 质粒及重组菌株的构建
利用CRISPR/Cas9基因编辑系统进行基因整合和启动子替换[18]。以将PthrABC替换为PBBa_J23108为例。利用sgRNA设计工具CRISPR RGEN Tools(http://www.rgenome.net/cas-designer/)设计20 bp间隔序列(pGRB-thrABC-1和pGRB-thrABC-2),退火后利用一步法克隆试剂盒与线性pGRB连接,转化E. coliDH5α并筛选后获得质粒pGRBPthrB。以E. coliW3110基因组DNA为模板,分别利用引物thrL-U-F/PBBa_J23108-U-R和PBBa_J23108-D-F/thrL-D-R扩增并通过重叠PCR获得含PthrABC上下游同源臂和PBBa_J23108的供体DNA。将PthrB(100 ng)和供体DNA(200 ng)电转化至含pREDCas9的E. coliW3110ΔlacI感受态细胞中。复苏2 h后,涂布于含100 μg/mL的氨苄青霉素和奇霉素的LB固体培养基上,于32 ℃培养过夜。挑选单菌落,利用引物thrL-U-F/thrL-D-R进行菌落PCR鉴定,将鉴定正确的转化子于含10 mmol/L阿拉伯糖的LB液体培养基中32 ℃振荡培养过夜,以丢失pGRB-PthrB。利用菌落PCR对重组菌株进行鉴定,并将PCR产物委托苏州金唯智生物科技有限公司测序验证。菌株于LB液体培养基42 ℃振荡培养过夜以丢失pREDCas9质粒。每次构建获得的菌株于LB液体培养基活化并连续传代10 次,稀释后涂布于LB固体培养基,挑取3~5 个单菌落进行摇瓶发酵实验,选取L-高丝氨酸产量高且生物量无显著降低的菌株进行后续改造。
以E. coliW3110基因组DNA为模板,利用引物rhtA-1和rhtA-2扩增rhtA基因,PCR产物回收后连接至经NcoI酶切的pTrc99a质粒,经转化、筛选、菌落PCR鉴定后获得重组质粒pTrc99a-rhtA。将pTrc99a-rhtA电转化至H-9,经筛选、菌落PCR鉴定后获得菌株H-10。
1.3.2 发酵实验
1.3.2.1 摇瓶发酵
将适量经斜面培养基活化后的菌体细胞接种至含30 mL种子培养基的500 mL摇瓶中,35 ℃、220 r/min振荡培养10 h。以1%接种量接种至45 mL发酵培养基,35 ℃、220 r/min振荡培养48 h。发酵过程中根据情况补充80%葡萄糖,用氨水调节pH值约为7.0。利用H-10发酵时,接种后加入终浓度为0.05 mmol/L的异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)。
1.3.2.2 5 L发酵罐发酵
将适量经斜面培养基活化后的菌体细胞接种至装有2.5 L种子培养基的5 L发酵罐,流加氨水调节发酵液pH 6.8~7.2,溶氧维持20%~40%,通风量2~4 m3/h,搅拌转速200~800 r/min,35 ℃培养6 h。以10%接种量将种子培养物接至装有3 L发酵培养基的5 L发酵罐进行发酵培养,发酵温度35 ℃,通风量2~4 m3/h,搅拌转速300~900 r/min,溶氧维持在20%~50%,流加80%的葡萄糖溶液,维持残糖质量浓度为1~3 g/L,流加氨水调节发酵液pH 6.8~7.2,发酵周期52 h。利用H-10发酵时,接种后加入终浓度为0.05 mmol/L的IPTG。
1.3.3 检测与分析方法
用生物传感器分析仪测定葡萄糖含量。生物量以OD600nm计。采用高效液相色谱柱前衍生法测定L-高丝氨酸浓度。样品用2,4-二硝基氟苯衍生,以乙腈-水混合液(1∶1,V/V)和50 mmol/L乙酸铵溶液为流动相,以1 mL/min的流速梯度混合经过C18AAA色谱柱分离,最后在360 nm波长处进行检测[19]。
2 结果与分析
2.1 弱化苏氨酸操纵子thrABC对L-高丝氨酸合成的影响
目前报道的常用策略是通过敲除高丝氨酸激酶编码基因thrB阻断L-高丝氨酸的降解,实现其积累。与阻断策略不同,适度弱化降解途径不会引起菌株营养缺陷,故可在不影响菌株生长的前提下实现代谢产物的积累[15,19]。在E. coli中,高丝氨酸激酶编码基因thrB与苏氨酸合成酶编码基因thrC及天冬氨酸激酶/高丝氨酸脱氢酶编码基因thrA组成操纵子thrABC,而高丝氨酸激酶和苏氨酸合成酶均参与L-高丝氨酸的降解[20-21]。为不破坏thrABC操纵子的天然结构并实现thrB和thrC的弱化,拟将该操纵子的启动子替换为不同强度的PBBa_J23108、PBBa_J23109和PBBa_J23114。但启动子替换会引起L-高丝氨酸关键合成基因thrA的弱化,故先在出发菌株E. coliW3110 ΔlacI基因组先整合1 个拷贝强启动子Ptrc调控的thrAC1034T(解除L-苏氨酸反馈抑制)[22]。利用鉴定引物ylbe-U-F/ylbe-thrA-J进行菌落PCR鉴定,thrAC1034T基因整合菌株基因组DNA扩增获得碱基约为1 013 bp的条带(图2),表明thrAC1034T基因整合成功,将菌株命名为H-1。在H-1的在整个发酵过程中,未检测到L-高丝氨酸积累(图3A),其原因可能是L-高丝氨酸被代谢为L-苏氨酸和L-甲硫氨酸等代谢物。
为弱化L-高丝氨酸降解,将H-1的PthrABC分别替换为PBBa_J23108、PBBa_J23109和PBBa_J23114。由出发菌株H-1基因组DNA应扩增出碱基数为1 599 bp的条带,而启动子替换菌株基因组DNA扩增获得碱基约为1 131 bp的条带,由图4可知,启动子替换成功,将菌株分别命名为H-2、H-3和H-4。为考察PthrABC替换对菌株生长和L-高丝氨酸合成的影响,对其进行摇瓶发酵实验。在不添加L-苏氨酸的培养基中H-2、H-3和H-4能够生长,表明PthrABC替换为弱启动子未造成其L-苏氨酸缺陷(图3B);H-2无L-高丝氨酸积累,H-3和H-4可分别合成1.5 g/L和0.8 g/LL-高丝氨酸(图3A)。PBBa_J23108、PBBa_J23114和PBBa_J23109的转录强度依次降低,PBBa_J23108分别是PBBa_J23114和PBBa_J23109的约5 倍和50 倍。由此推测,因PBBa_J23108转录强度较高不利于L-高丝氨酸的积累;PBBa_J23114和PBBa_J23109有效降低了thrBC的转录,L-高丝氨酸得以积累;又因PBBa_J23109转录强度低于PBBa_J23114,故H-3的L-高丝氨酸产量高于H-4。此外,由图3B可知,与对照菌株H-1相比,H-3和H-4最终生物量有所降低,且H-3生物量低于H-4,与菌株L-高丝氨酸产量呈相反趋势,其原因可能是thrBC转录的下调有效弱化了菌株生长代谢流。Li Hua等[16]敲除thrB并过表达thrA,菌株L-高丝氨酸产量达到1.2 g/L,较H-3低20%,且在发酵过程需添加L-苏氨酸。综上,适度弱化thrBC转录可实现L-高丝氨酸的积累,且不会造成营养缺陷,其效果优于敲除thrB。

图2 菌株H-1鉴定图谱Fig. 2 Identification of strain H-1

图3 PthrABC替换对菌株生长和L-高丝氨酸合成的影响Fig. 3 Effect of thrABC promoter substitution on the growth of strains and L-homoserine production
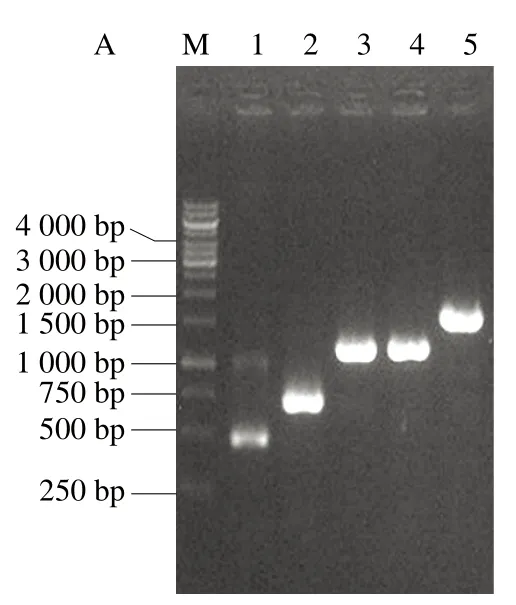

图4 菌株H-2(A)、H-3(B)和H-4(C)鉴定图谱Fig. 4 Identification of strains H-2 (A), H-3 (B) and H-4 (C)
2.2 增强thrA拷贝数对L-高丝氨酸合成的影响
尽管通过弱化降解途径实现了L-高丝氨酸的积累,但其产量较低。天冬氨酸激酶是L-高丝氨酸合成的关键酶[20-21]。在E. coli中共含有3 个天冬氨酸激酶I、II和III,其中thrA的天冬氨酸激酶I丰度最高,在合成L-高丝氨酸中起主要作用[22]。为进一步提高L-高丝氨酸的合成代谢流,向H-3再整合1 拷贝的thrAC1034T(受强启动子Ptrc调控)。由图5A可知,thrAC1034T基因整合成功,将其命名为H-5。摇瓶发酵实验结果表明,H-5的L-高丝氨酸产量为2.6 g/L,较H-3提高73.3%(图6)。为考察进一步过表达thrAC1034T对高丝氨酸合成的影响,向H-5再整合1 拷贝的thrAC1034T(受强启动子Ptrc调控)。由图5B可知,thrAC1034T基因整合成功,将其命名为H-6。摇瓶发酵实验结果表明,H-6的L-高丝氨酸产量为3.4 g/L,较出发菌株提高30.7%。

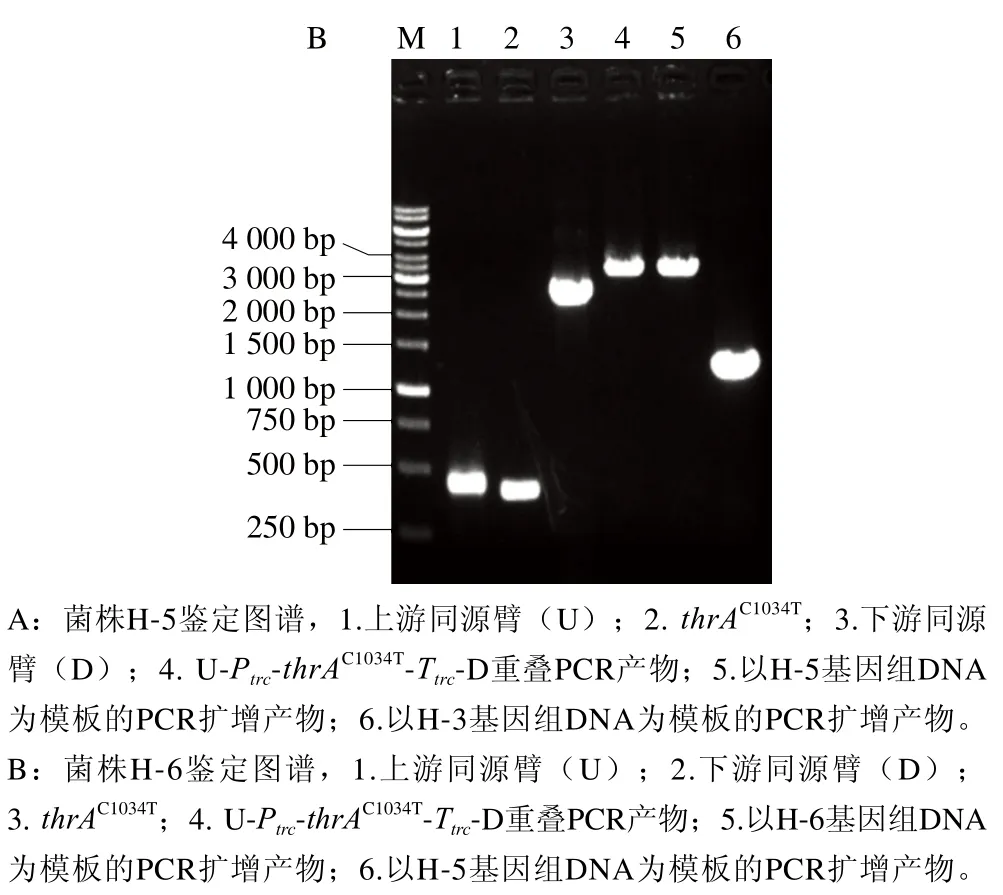
图5 菌株H-5和H-6鉴定图谱Fig. 5 Identification of strains H-5 and H-6
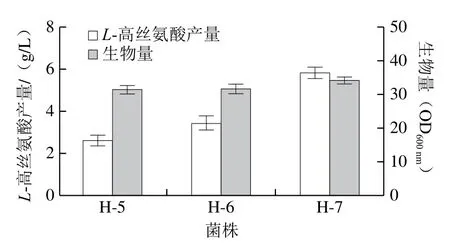
图6 菌株H-5、H-6和H-7的L-高丝氨酸产量和生物量Fig. 6 L-Homoserine production and biomass of strains H-5, H-6 and H-7
2.3 增加草酰乙酸合成对L-高丝氨酸合成的影响

图7 菌株H-7鉴定图谱Fig. 7 Identification of strain H-7
由图6可知,在H-6的L-高丝氨酸产量提高程度较H-5低,推测L-高丝氨酸前体物供应不足限制了其合成。草酰乙酸是合成包括L-高丝氨酸在内的L-天冬氨酸族氨基酸的重要前体物[19,23-25]。为增加L-高丝氨酸合成代谢流,向H-6基因型整合1 拷贝由自身启动子调控的磷酸烯醇式丙酮酸羧化酶编码基因ppc,鉴定结果如图7所示,由出发菌株H-6基因组扩增出碱基数为1 873 bp的条带,由ppc基因整合菌株基因组扩增获得碱基约为3 000 bp的条带,与预期一致(3 249 bp),表明菌株构建成功,将其命名为H-7。H-7的L-高丝氨酸产量达5.8 g/L,比菌株H-8提升了70.5%(图6)。上述结果表明,草酰乙酸是L-高丝氨酸的限制因素,提高其供应有利于增强L-高丝氨酸的合成。
2.4 增加NADPH再生对L-高丝氨酸的影响

图8 菌株H-8和H-9鉴定图谱Fig. 8 Identification of strains H-8 and H-9
在L-高丝氨酸生物合成途径中,高丝氨酸脱氢酶和天冬氨酸半醛脱氢酶催化的反应需要NADPH。生成1 molL-高丝氨酸需要2 mol NADPH,由此推测NADPH供应是L-高丝氨酸的另一限制因素。研究证明,过表达吡啶核苷酸转氢酶编码基因可有效促进NADPH的再生并增强L-赖氨酸、L-缬氨酸等代谢物合成[11,26-28]。为增强NADPH的再生,分别将自身启动子PpntAB和Ptrc调控的pntAB整合至H-7基因组。由菌株H-7基因组DNA应扩增出碱基数为1 681 bp的条带,由pntAB整合菌株基因组DNA扩增获得碱基可扩增出4 443 bp(PpntAB-pntAB)和3 994 bp(Ptrc-pntAB)的条带。由图8可知,pntAB基因整合成功,将菌株分别命名为H-8和H-9。摇瓶发酵结果如图9所示,H-8的L-高丝氨酸产量(7.8 g/L)略高于H-9(7.1 g/L),说明pntAB自身启动子效果略优于Ptrc。上述结果表明,增强NADPH再生可有效促进L-高丝氨酸的合成。以往研究均聚焦于L-高丝氨酸代谢途径的改造,鲜见增强还原力供应的相关报道[4,8-9,16-17]。本研究证明了强化pntAB表达可显著提高L-高丝氨酸的合成。

图9 菌株H-8、H-9和H-10的L-高丝氨酸产量和生物量Fig. 9 L-Homoserine production and biomass of strains H-8,H-9 and H-10
2.5 促进L-高丝氨酸输出对其产量的影响
胞内高浓度的L-高丝氨酸积累可抑制谷氨酸脱氢酶的活性,从而对菌体细胞生长具有毒害作用[29]。由图7、9可知,H-8和H-9的生物量较H-7显著降低,可能与L-高丝氨酸的毒害作用有关。L-苏氨酸/L-高丝氨酸输出载体具有外排L-高丝氨酸的功能,过表达其编码基因rhtA可有效提高L-高丝氨酸的产量和菌体生物量[16-17,30]。为增强L-高丝氨酸输出以提高其产量和菌株抗性,构建了rhtA过表达质粒pTrc99a-rhtA,并将其转化至H-8,获得菌株H-10。如图9所示,H-10的L-高丝氨酸产量达12.5 g/L,比H-8提高了60.2%;其生物量提高12.6%,表明增强L-高丝氨酸输出可有效提高其产量并增强菌株对L-高丝氨酸的抗性。
2.6 发酵罐发酵实验
为进一步考察H-10的发酵性能,于5 L发酵罐进行发酵实验。如图10所示,菌株于8 h进入对数生长期,32 h后进入稳定期,随后生物量缓慢下降;于4 h可检测到L-高丝氨酸的积累,12 h后合成速率逐渐提升,48 h达到最高,52.1 g/L,其转化率为43%(以占葡萄糖质量计)。由表3可知,本研究所构建的H-10L-高丝氨酸合成效率高于现有报道且该菌株无营养缺陷,故无需在发酵过程中额外添加营养物质,具有工业化生产的优势。
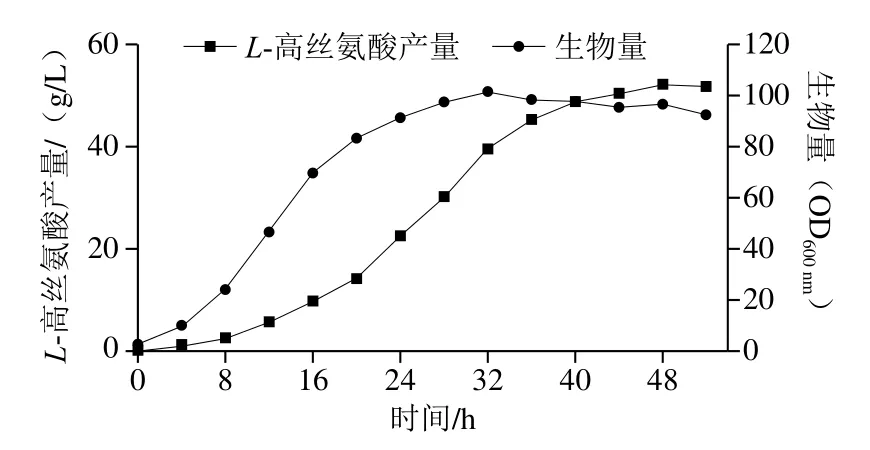
图10 H-10发酵过程曲线Fig. 10 Time courses of L-homoserine production and biomass of strain H-10

表3 本研究与报道文献的参数比较Table 3 Comparison of L-homoserine production performance between strain H-10 and E. coli
3 结 论
采用系统代谢工程策略构建了1 株无营养缺陷的L-高丝氨酸高产菌株H-10,主要策略包括利用弱启动子调控thrB和thrC转录弱化L-高丝氨酸的降解代谢流;过表达关键酶基因thrAfbr、ppc和pntAB,增强其合成代谢流、前体物草酰乙酸供应及NADPH再生;过表达L-高丝氨酸输出蛋白增强其外排。于5 L发酵罐经发酵48 h,H-10菌株的L-高丝氨酸产量达到52.1 g/L,高于现有报道且发酵过程中无需额外添加营养物质,故具备工业化潜力。本研究采用的代谢途径弱化策略可为工业微生物的代谢工程构建提供借鉴。
