聚合物原位成纤方法及其在PP共混体系中的应用
2022-03-25王富玉郭金强张玉霞
王富玉,郭金强,张玉霞*
(北京工商大学化学与材料工程学院,北京 100048)
0 前言
目前聚合物材料的应用越来越广泛,但通用塑料的力学性能等不能满足工程与结构件更高性能使用要求[1]。共混改性是提高聚合物基体性能的重要途径。其中一种方法是通过添加高强度材料如玻璃纤维、碳纤维等来提高聚合物的拉伸强度、弹性模量等[2],但纤维会导致体系的黏度增加、设备磨损大,且回收困难[3];另一种方法是通过改变加工工艺条件,调控两相共混物中分散相在连续相中的形态,进而提高聚合物材料的性能,如采用聚合物原位成纤方法制备原位成纤共混体系。
“原位微纤复合材料”这一概念是由Kiss和Weiss等在1987年先后提出的[4-5]。原位成纤是指将聚合物共混后,通过施加外力场如拉伸场或剪切场使共混体系中的分散相受到拉伸或剪切作用而沿外力方向取向,并且相形态微纤化,在连续相中形成取向微(纳)纤,进而实现对连续相增强[6]。与传统纤维增强相比,聚合物原位成纤所形成的微纤具有与连续相相容性好、分散均匀、增强效果明显且可控、可回收利用等优点[3],近年来对原位成纤共混体系的研究逐渐增多。
1 聚合物共混体系原位成纤原理及影响因素
1.1 原位成纤原理
从流变学角度分析,原位成纤是聚合物在加工过程中受热熔融,熔体中分散相“液滴”在挤出过程中受到螺杆剪切作用或在口模区受到拉伸作用,产生较大形变,形成棒状、短粗微纤或长径比大的微纤,如图1[7]所示。分散相微纤在连续相中起到增强骨架的作用;还可以起到异相成核作用,使连续相沿着微纤结晶或与微纤形成串晶结构,改善材料界面应力分布和传递,进而提高聚合物材料的使用强度[8]。

图1 原位成纤形成原理示意图[7]Fig.1 Schematic diagram of in-situ fibrillation process[7]
1.2 原位成纤影响因素
影响聚合物共混体系原位成纤的因素有很多,大致可分为内因和外因两方面,其中内因主要是共混物中分散相的用量与性能,包括其与连续相的共混比、黏度比、相容性等;外因主要是加工工艺,包括加工温度、拉伸速率(拉伸比)、螺杆转速等[9]。
1.2.1 内因
(1)共混比
共混体系中分散相含量较少时,液滴小,难以成纤或形成短纤;分散相含量高时,易团聚或形成较大椭球状液滴。Li等[10]研究了聚对苯二甲酸丙二醇酯/乙酸-丁酸纤维素(PTT/CAB)(质量比分别为10/90、20/80/、30/70、40/60、50/50)原位成纤共混体系(图2),发现在PTT含量低于30%(质量分数,下同)时,形成了微纳纤;但随着PTT含量增加,微纤变粗,直径分布变宽;当PTT含量为40%时,分散相之间呈片层连接结构;当PTT含量为50%时,呈现共连续结构,不能形成微纤。

图2 不同配比的PTT/CAB共混物中PTT分散相的形貌[10]Fig.2 Morphology of PTT phase in PTT/CAB polymer blends with different blending ratios[10]
董珈豪等[11]采用微纳叠层共挤出工艺制备了乙烯-辛烯共聚物/PTT(POE/PTT)原位成纤共混体系,发现PTT含量为5%时,大多数PTT呈颗粒状、椭球状、短微纤状,只有少量PTT成纤;随着PTT含量增加,PTT成纤增加,长径比增大;拉伸强度沿挤出方向先增加后降低。PTT含量为15%时,拉伸强度沿挤出方向达到最大值;PTT含量为20%时,微纤最多,但微纤与基体的弱界面作用产生大量空穴,不利于增强PP,拉伸强度沿挤出方向降低。
因此,对不同共混体系应探求其最佳共混比,以使分散相成纤效果最佳,进而使连续相性能得到最大程度改善。
(2)黏度比
共混体系中分散相与连续相之间的黏度比(ηd/ηm≈λ)对不同流场作用下的分散相液滴破裂及分散相粒径有着显著影响[12],研究表明,λ与分散相形态的关系如下:λ<0.7时,分散相为微纤状;0.7<λ<1.7时,分散相为微纤与颗粒共存状态;λ>2.2时,分散相为颗粒状[2]。
刘渝等[13]研究了不同黏度比对PP在EVA中成纤状态的影响,结果发现,如图3所示。当黏度比为0.49和0.53时,分散相PP为棒状微纤,直径较大,且分布较宽,长径比较小;黏度比为0.66时,分散相PP为微纤状,直径较小且均匀,微纤长径比大,易缠结成微纤网络结构。

图3 不同黏度比时EVA/PP共混物中PP微纤的形貌[13]Fig.3 Morphology of PP microfibers in EVA/PP blends with different viscosity ratios[13]
张玲等[14]制备了2种不同黏度比的POE/PP原位成纤共混体系,研究了黏度比对POE在PP基体中微纤形态的影响,发现黏度比<1时,POE更易形成微纤,微纤长径比较大,微纤较多;黏度比>1时,POE也能形成微纤,但微纤长径比较小,微纤较少。尽管该共混体系在黏度比>1时也可以成纤,但不如黏度比<1时的成纤效果好。
(3)相容性
有研究表明,要形成较好的原位成纤共混体系,要求共混物两相处于弱相容性或适中的相容性,在外力作用下分散相与连续相之间才能产生滑移并发生形变,形成微纤。
易新等[15]研究了相容剂对PET在PP中分散成纤的影响。结果表明,未加相容剂的共混体系,微纤具有较大长径比,微纤直径分布均匀;而加入相容剂后微纤长径比减小,微纤直径分布不均匀。因此,PP/PET原位成纤共混体系中分散相PET微纤的形态与相容剂密切相关。
贾世奎等[16]在聚乳酸/聚丁二酸丁二醇酯(PLA/PBS)共混物中添加过氧化二异丙苯(DCP)(添加量分别为0.1%、0.3%、0.5%、1.0%、1.5%)制备了PLA/PBS/DCP(PLA与PBS/DCP质量比为10/90)原位成纤共混体系,发现DCP的加入使PLA和PBS两相界面间隙减小,相容性提高;随着DCP含量增加,微纤直径减小;当DCP含量为1%时,PLA微纤直径达0.2~0.4 μm;DCP含量为1.5%时,PLA界面相容性好,微纤消失。
赵均乐等[17]在线形低密度聚乙烯(PE-LLD)/PET(80/20)中添加不同含量相容剂线形低密度聚乙烯与甲基丙烯酸缩水甘油酯接枝物(PE-LLD-g-GMA,用量分别为2、5、10份)制备了不同的原位成纤共混体系,发现添加2份相容剂时,共混体系断面有少量细长微纤及大量短微纤和球状粒子,且微纤分布不均匀;添加5份和10份相容剂时,共混体系断面平整,微纤很少,长径比变小,出现更多长径比小的短微纤和球状粒子。
由此可见,只有两相相容性适宜时,分散相才能形成微纤。
1.2.2 外因
(1)加工温度
一般来说,聚合物两相共混原位成纤要求分散相的熔融温度(T2)高于连续相的熔融温度(T1)至少30℃以上,即T2-T1>30℃,且拉伸成纤时要求加工温度T在两相熔融温度之间,即T2>T>T1[10]。
王占杰[18]在2个不同注塑温度下制备了均聚聚丙烯(PP-H)/PET(85/15)原位成纤共混体系(表2),低温注塑样品注塑温度为220℃,低于PET熔点(248℃);高温注塑样品注塑温度为280℃,高于PET熔点,结果发现高温注塑试样力学性能低于低温注塑试样。原因是低温注塑时温度低于PET的熔点,样品中PET微纤可较好地保留,起到增强作用;而高温注塑时温度偏高时,制样时使PET微纤重新熔融回缩,以球状或椭球状的形态分布在基体中,不能起到增强作用。
黄英等[19]研究了加工温度对PP/PA66(90/10、80/20、70/30)共混体系中PA66微纤直径和长径比的影响(图4),发现在245℃左右时,PA66微纤直径最小,长径比最大。而当温度较低时,PA66刚性大,变形能力弱,回缩性强,难以成纤;当温度接近PA66熔点(263℃)时,PA66基本处于熔融态,易变形且易恢复形变,微纤不能稳定存在于连续相PP中。
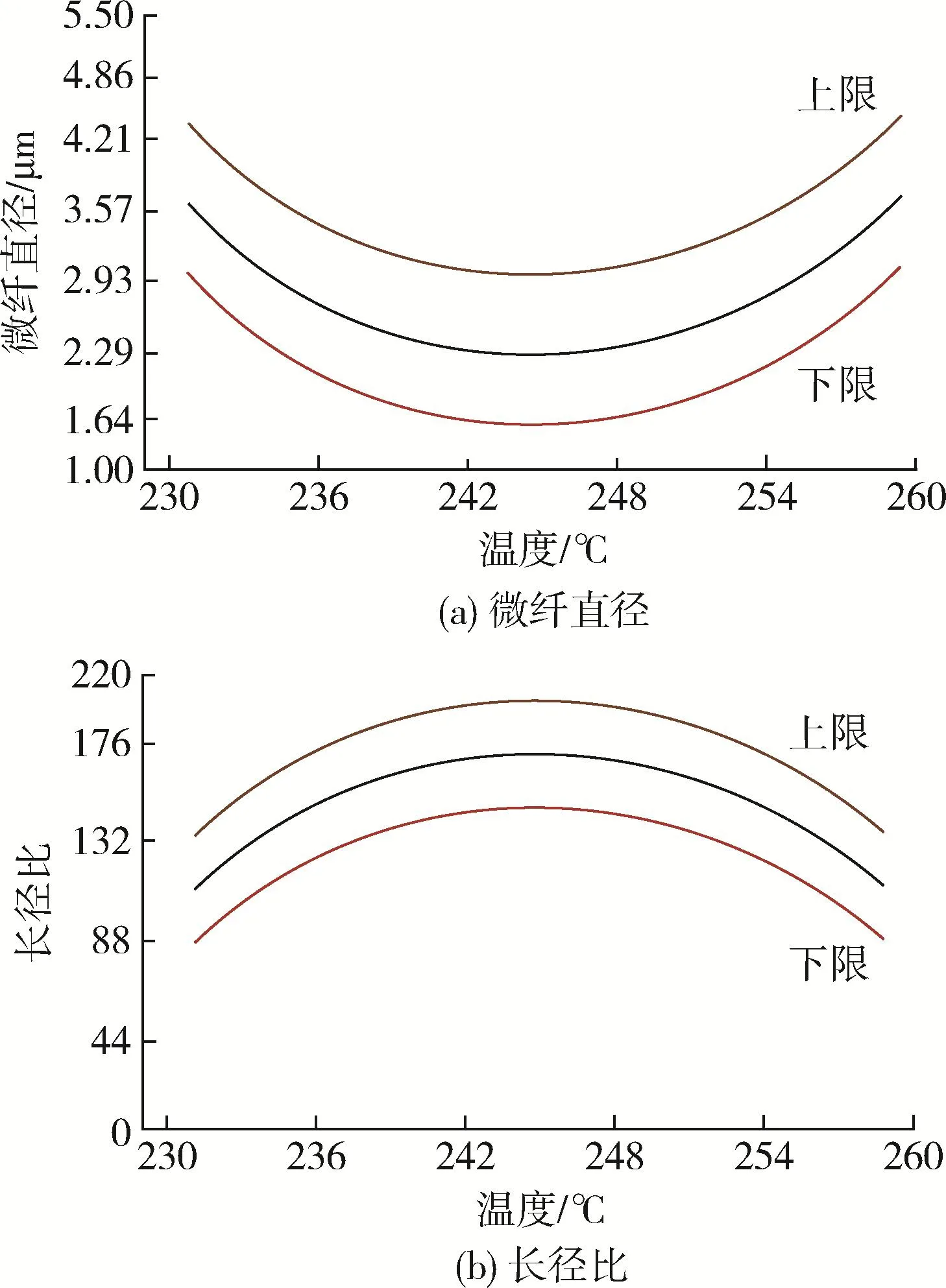
图4 加工温度对PP/PA66共混体系中PA66微纤直径和长径比的影响[19]Fig.4 Effect of processing temperature on diameter and aspect ratio of PA66 microfiber in PP/PA66 blends[19]
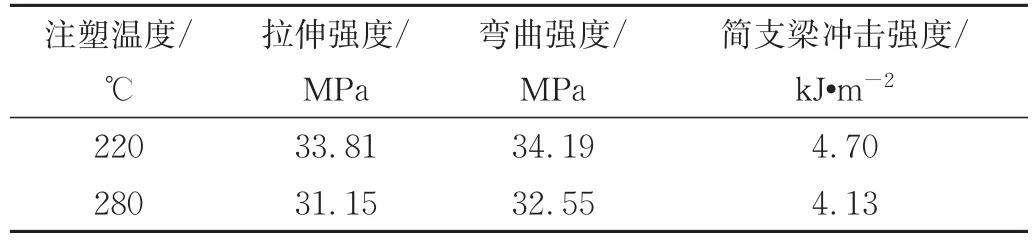
表1 注塑温度对PP-H/PET共混体系力学性能的影响[18]Tab.1 Influence of injection molding temperature on mechanical properties of PP-H/PET blends[18]
因此适宜的加工温度对于PA66在PP中的成纤至关重要。
(2)拉伸速率(拉伸比)
拉伸作用小时,难以成纤;拉伸作用大时,微纤易拉断[20]。Sun[21]通过多级挤出设备制备了质量比为80/20的乙烯-辛烯共聚物(POE)/PLA原位成纤共混体系,研究了拉伸速率对成纤体系性能的影响。结果表明,随着拉伸速率由40、50、60 r/min增加到70 r/min时,PLA微纤增多,但直径由0.76 μm减小至0.66 μm后增大到0.68 μm,且直径分布变窄;在拉伸速率为60 r/min时,PLA微纤直径为0.66 μm,且具有最大的长径比。
李忠明等[20]采用熔融挤出—热拉伸—淬冷的方法制备了PET/PE(85/15)原位成纤共混体系,发现热拉伸比较小时,分散相PET为棒状,且棒长短、粗细不均匀;拉伸比较大时,分散相PET为微纤状,且微纤直径比较均匀。
任曙霞[22]采用同样的方法制备了PE/PTT/PE-g-MAH(70/30/4)原位成纤共混体系,发现随着拉伸速率增加,PTT由液滴状变成微纤,且数量增多,直径变小,同时微纤粗细趋于均匀,如图5所示。

图5 不同拉伸速率时PE/PTT原位成纤共混体系的SEM照片[22]Fig.5 SEM photos of PE/PTT in-situ fibrillation blends with different drawing rate[22]
(3)螺杆转速
螺杆转速决定剪切速率,剪切速率高有利于分散相成纤和均匀分布;剪切速率低不利于分散相成纤和均匀分布。孙永锋等[23]制备了高密度聚乙烯(PEHD)/PA6(90/10)原位成纤共混体系,发现不同螺杆转速时体系中PA6微纤数量和分布不同。螺杆转速为20 r/min时,微纤少,且分布也不均匀;螺杆转速为50 r/min时,形成的微纤较多且分布均匀。
Li等[7]通过挤出拉伸制备了烯烃嵌段共聚物(OBC/PP)(质量比分别为90/10、95/5、85/15、80/20)原位成纤共混体系,研究了螺杆转速对PP成纤的影响。结果表明,在高拉伸比时,螺杆转速越高,PP微纤直径越小;低拉伸比时,螺杆转速越低,微纤直径越大,长径比越小,PP为椭球状短微纤。随着体系中PP形态实现球状—椭球状—短微纤状—连续微纤状的转变,共混体系拉伸强度和模量逐渐增加。
2 原位成纤制备方法
聚合物共混体系中分散相需受外力场作用才能形变成微纤。原位成纤有不同的制备方法,如熔融挤出—热拉伸—淬冷[6]、熔融挤出—冷拉伸—退火[16]、微纳叠层共挤出成型[24-25]、多级拉伸成型[26]、循环震荡推拉成型(LOPPM)[6]、动态成型保压成型(DPIM)[27]、强剪切机头挤出成型[28]等。
2.1 熔融挤出—热拉伸—淬冷工艺
熔融挤出—热拉伸—淬冷工艺是目前较为广泛采用的一种方法,是将2种聚合物进行熔融共混挤出后直接对熔体进行热拉伸,使分散相形态变为微纤状,之后再对聚合物共混体系淬冷,将分散相微纤保留在连续相中,得到原位成纤共混体系(见图6)。其优点是制备过程连续,工艺可控[6]。
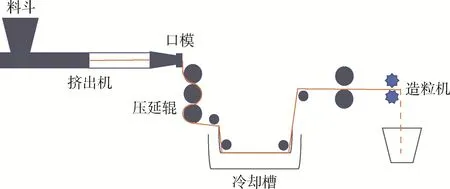
图6 熔融挤出—热拉伸—淬冷工艺制备原位成纤共混体系示意图[23]Fig.6 Schematic of molten extrusion-hot drawing-quenching process for the preparation of in-situ fibrillation[23]
刘欢等[29]利用螺杆头部有直槽混炼件的单螺杆挤出机,分别采用熔融直接挤出和熔融挤出—热拉伸—淬冷2种工艺制备了PP/PET(80/20)原位成纤共混体系,发现直接熔融挤出制备的共混体系中分散相PET为球状和少量短粗微纤;而熔融挤出—热拉伸—淬冷工艺制备的共混体系中分散相PET为大量细长微纤,长径比超过20时,微纤直径约为5 μm,如图7所示。

图7 不同工艺制备的PP/PET原位成纤共混体系SEM照片Fig.7 SEM photos of PP/PET in-situ fibrillation blends prepared by different processing methods
2.2 熔融挤出—冷拉伸—退火工艺
熔融挤出—冷拉伸—退火工艺(见图8)是将2种聚合物熔融共混后冷却,对体系进行冷拉伸,使两相都发生取向。之后在分散相熔点以下、连续相熔点以上进行退火处理,使连续相分子链解取向,而分散相维持原形态不变。这种方法对2种聚合物的黏弹性和熔点有一定的要求,退火时连续相回缩而分散相保持原状,退火温度须高于连续相温度而低于分散相温度。该工艺较为复杂[6],且退火后分散相成纤效果会受到影响。
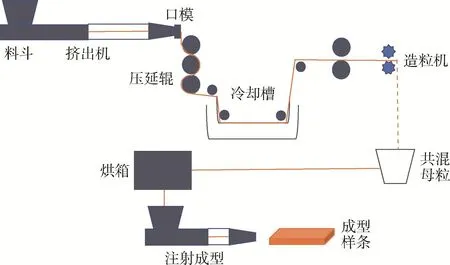
图8 熔融挤出-冷拉伸-退火工艺原位成纤共混体系示意图[6]Fig.8 Schematic of molten extrusion-cold drawing-annealing process for the preparation of in-situ fibrillation[6]
赵均乐等[17]采用这一工艺制备了PE-LLD/PET(80/20)原位成纤共混体系,发现退火后,分散相PET微纤分布不均匀、取向不一致且变短,如图9所示。

图9 退火前后的LLDPE/PET原位成纤共混体系SEM照片[17]Fig.9 SEM photos of LLDPE/PET in-situ fibrillation blends before and after annealing[17]
2.3 微纳叠层共挤出工艺
Schrenk[30]首次提出微纳叠层共挤出方法,将2种或2种以上具有不同性能的聚合物通过2台挤出机分别熔融塑化后流入具有特殊结构的层叠单元(倍增器),经过倍增器的多次分割和叠合,最终制备出厚度可达微纳级的多层共混体系[31-33]。
Shen等[34]利用多层共挤出装置制备了PA6/iPP(95/5)原位成纤共混体系,发现随着倍增器层数的增多,分散相PA6趋向于沿着熔体流动方向成纤。与未使用倍增器的共混体系相比,用9层倍增器挤出的共混体系拉伸强度由19 MPa提高到47 MPa,断裂伸长率提高了100%。
李婷等[35]利用微纳叠层共挤出方法将PP与EVOH(PP/EVOH=80/20)共混体系沿挤出方向交替排列,使EVOH在PP连续相中的分散形态由零维球形变为一维微纤状(图10)。

图10 PP/EVOH共混体系平行挤出方向SEM照片[35]Fig.10 SEM micrographs of PP/EVOH blends parallel to extrusion direction[35]
2.4 多级拉伸工艺
在多级拉伸设备上设置分割叠加单元(LME)制备原位成纤共混体系时,在聚合物熔体经过分割叠加单元时,被均匀地分割成两部分,分别进入分割的流道,最后这两部分被分开的熔体在口模出口处在垂直方向上重新叠合在一起。与传统基于剪切力场的挤出装置相比,多级拉伸设备具有更好的分散效果及取向能力,在调控分散相形态与聚合物晶体结构、促进纳米填料分散等方面具有突出的优势[26]。

图11 多级双向拉伸系统示意图[26]Fig.11 Schematic of multistage biaxial stretching extrusion system[26]
孙小杰等[36]采用设置了LME的多级拉伸装备制备了 PP/PA1010/碳黑(PP/PA1010/CB=90/8/2)原位成纤共混体系,研究了不同数量LMEs对PA1010成纤形态的影响。发现无LME时,分散相PA1010呈球状或椭球状;设置3个LMEs时,分散相PA1010为大长径比微纤;设置5个LMEs时,分散相PA1010微纤部分被拉断成更短的微纤。

图12 PP/PA1010/CB共混体系沿挤出方向SEM图(×2 000)[36]Fig.12 SEM micrographs of PP/PA1010/CB blends along the flow direction(FD)(×2 000)[36]
3 原位成纤方法在PP共混体系中的应用
PP是一种综合性能优良的热塑性塑料,但拉伸强度等尚不能满足高性能工程应用如汽车、工程结构件等的要求,其高性能化一直是研究热点。采用原位成纤方法制备PP基原位成纤共混体系,对改善其微观结构、提高性能有明显效果,为实现其高性能化提供了一种解决方案。
3.1 PP/PA原位成纤共混体系
3.1.1 PP/PA6原位成纤共混体系
PA6具有拉伸强度高等特点,但与PP极性相差大,两者是不相容体系,因此要获得性能优异的PP/PA6共混物,最有效和使用最多的方法就是添加相容剂。孙义明等[37]研究了未加相容剂、MAH原位增容及PP-g-MAH增容对PP/PA6共混体系力学性能的影响,发现添加了相容剂的体系力学性能优于未添加体系。用PA6增强PP时,其易在PP基体中原位成纤,但需添加相容剂制备PP/PA6原位成纤共混体系[38]。
王玉等[39]以马来酸酐接枝PP(PP-g-MAH)为相容剂,采用熔融挤出—热拉伸法制备了一系列PP/PA6(85/15、84.5/15/0.5、83/5/15/1.5、82/15/3)原位成纤共混体系。研究表明,相容剂含量对微纤形态有明显影响,如图13所示。当PP-g-MAH含量为0.5%时,原位成纤共混体系中形成了大长径比的PA6微纤,其缠结结构和部分相界面的黏附使得原位微纤共混体系表现出较高的弹性响应,且其具有优异的异相成核作用,显著提高了共混体系的结晶度;同时PA6微纤的增强效应和相界面相容性的改善使得微纤共混体系的拉伸强度提高了15.49%;但PP-g-MAH含量继续增加,不利于共混体系中分散相PA6的变形,成纤效果变差,拉伸强度下降。

图13 不同相容剂含量PP/PA6/PP-g-MAH原位成纤共混体系的SEM照片[39]Fig.13 SEM of PP/PA6/PP-g-MAH blends with different content of compatibilizer[39]
孙显茹等[40-42]采用挤出—热拉伸—淬冷法制备了PP-H/PA6(质量比为90/10、85/15)原位成纤共混体系,PA6微纤直径达到微米级;加入少量相容剂PP-g-MAH改善了PP-H和PA6的界面相容性,更有利于PA6微纤的形成,共混体系的拉伸强度、弯曲强度和冲击强度显著提高。此外,他们还研究了无规聚丙烯(PP-R)/PA(质量比为97/3、95/5)原位成纤共混体系,发现不加相容剂时,PA6微纤与PP之间有较多空洞,说明分散相与连续相的相容性差;添加相容剂后,PA6微纤增多,直径变小。
因此,欲获得性能优异的PP/PA6原位成纤共混体系,需优化相容剂的用量。
此外,拉伸作用对PP/PA6原位成纤共混体系也有较大影响。黎学冬等[43]研究了不同拉伸速率对PP/PA6(85/15)原位成纤共混体系的力学性能和PA6微纤直径的影响,结果如表2所示。随着拉伸速率由6 m/min增加到30 m/min时,PA6微纤直径由9.15 μm减小到2.15 μm;且拉伸强度随着拉伸速率的增加先增加后降低,在24 m/min时拉伸强度达到最大值;拉伸速率再升高,拉伸强度反而下降,这是因为拉伸作用太大时,易拉断微纤。

表2 不同拉伸速率时PP/PA6原位成纤共混体系力学性能和PA6微纤平均直径[43]Tab.2 Mechanical properties of the PP/PA6 in-situ fibrillation blends and the average diameter of PA6 microfibers against different drawing rate[43]
3.1.2 PP/PA66原位成纤共混体系
除PA6外,PA66也具有强度高、刚性好、抗冲击等优点,也常用于增强PP。黄英[19]采用双螺杆挤出机制备了PP/PA66(质量比分别为90/10、80/20、70/30)原位成纤共混体系,发现影响PA66微纤形态的关键因素是分散相的用量和加工温度(图14)。一方面,随着PA66含量增加,PA66微纤直径先减小后增加,长径比一直增大,在PA66含量为30%时长径比达到最大值;一方面,低加工温度(230℃)时,PA66形变能力弱、回缩性强;高加工温度(260℃)时,PA66微纤易形变,不稳定,因此在高于连续相熔融温度(230℃)、低于分散相PA66熔点(263℃)的适宜加工温度(245℃)时,微纤直径较小,长径比最大。在PA66含量为30%、中间温度245℃、高螺杆转速200 r/min时,PP/PA66共混体系的拉伸强度和冲击强度最高。说明对于PP/PA66而言,此时PA66易于成纤,对PP的增强效果也明显。
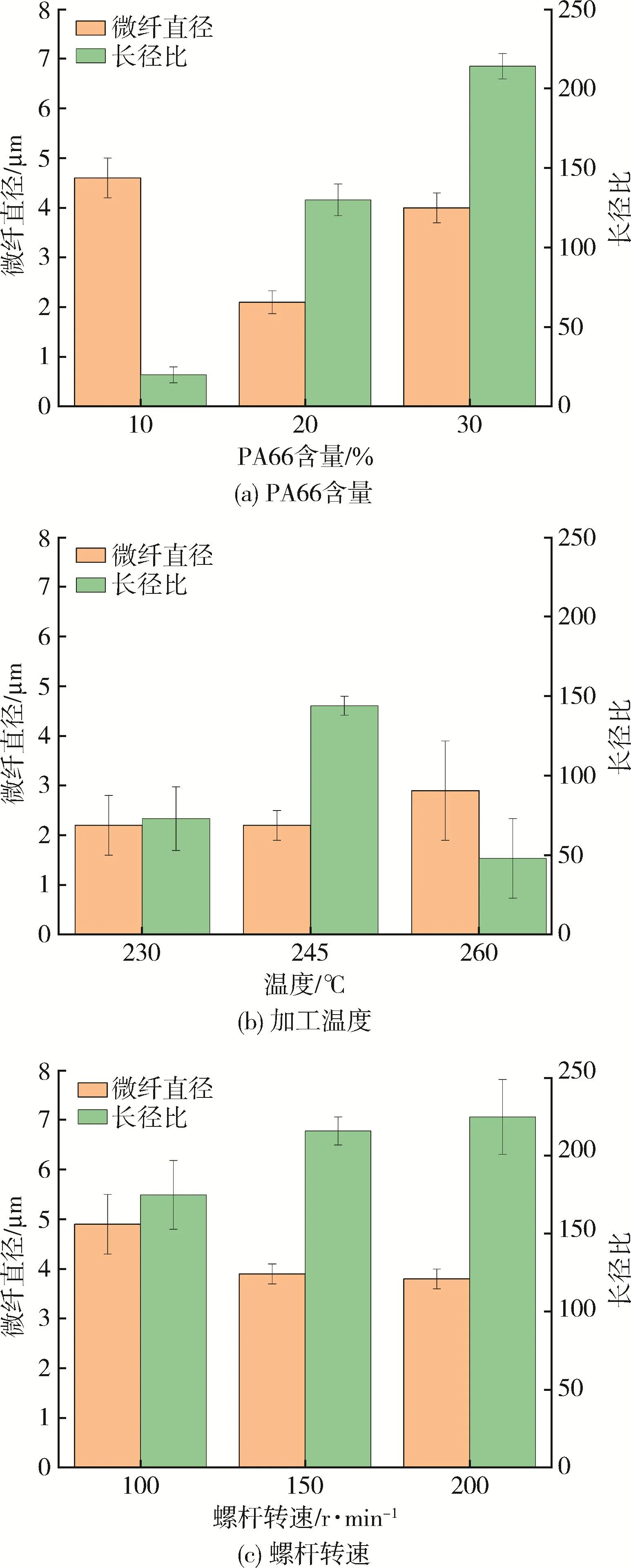
图14 不同工艺条件时PP/PA66中PA66微纤直径变化[19]Fig.14 The diameters of PA66 microfibrils in PP/PA66 blends under different processing conditions[19]
3.1.3 PP/PA11原位成纤共混体系
有研究表明,PA11在PP中也可成纤增强PP。戚远慧等[24]采用微纳叠层共挤出装置制备了PP/PA11(质量比分别为100/0、95/5、90/10、85/15、80/20、75/25)片材,发现随着PA11含量的增加,原位成纤共混体系中PA11微纤直径和PP结晶温度及结晶度先增加后减小。这是因为一定含量PA11可以作为PP的异相成核剂,提高PP结晶度;而当PA11含量超过25%时,反而不利于PP结晶。沿挤出方向共混体系拉伸强度随PA11含量的增加先增加后降低,而后又增加,在PA11含量为10%时,拉伸强度沿片材挤出方向达最大值25.4 MPa,比纯PP提高23.72%。
因此,在PP/PA原位成纤共混体系中,分散相PA的选择及其用量、加工温度、拉伸速率及相容剂等都会影响PA微纤形态,进而影响共混体系的力学性能和结晶性能等。
3.2 PP/PET原位成纤共混体系
PET具有强度高、模量高等特点,将其与PP共混制备原位成纤共混体系可提高PP的力学等性能,但两者相容性差,欲制备性能优良的PP/PET原位成纤共混体系,需优化相容剂的用量和工艺参数等。
孙显茹等[44]采用挤出—热拉伸—淬冷工艺制备了PPH/PET(质量比分别为 95/5、90/10、85/15、80/20、75/25)原位成纤共混体系,发现在合适的共混比和加工参数时,可形成PET微纤,且加入少量相容剂有利于PET微纤的形成。拉伸比由3∶1提高到20∶1时,共混体系拉伸强度由32.1 MPa提高到40.1 MPa,提高了24.9%。
Rizvi等[45]采用熔融共混—拉伸法制备了不同含量(1%、3%、5%、7%)PET的PP/PET原位成纤共混体系,发现分散相PET含量为3%时,PET呈现微纤网络结构,使得PP/PET共混体系在单向拉伸时应变硬化效果增强(图15)。这有利于改善其可发泡性等加工性能。
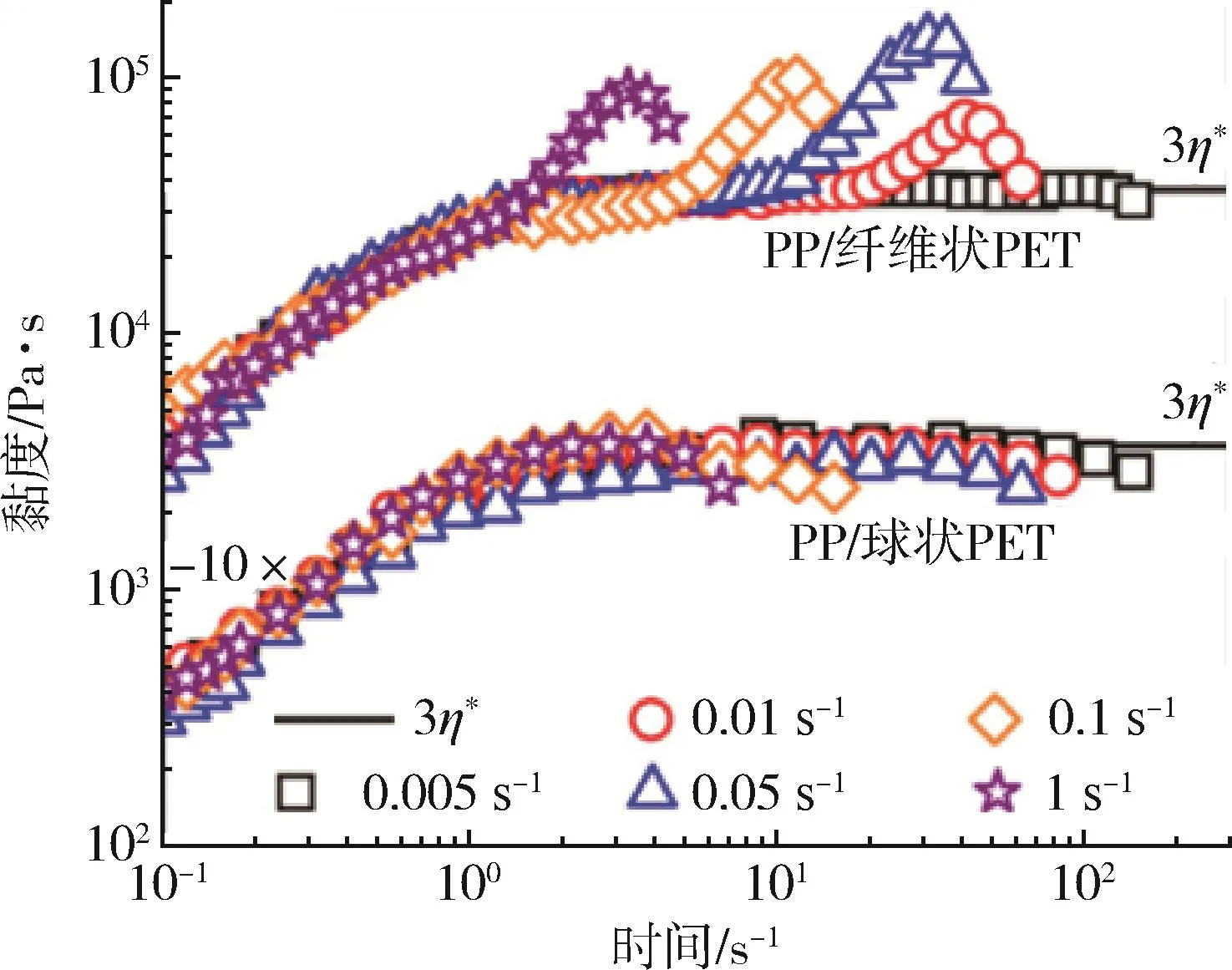
图15 测试温度为165℃时单向拉伸PP/PET(质量比为97/3)的黏度曲线[45]Fig.15 Viscosity of uniaxially oriented PP/PET measured at 165 ℃[45]
刘欢等[29]采用3种不同设备制备了PP/PET(质量比为80/20)原位成纤共混体系,发现采用双螺杆挤出机和配备三段式螺杆的单螺杆挤出机制备的PP/PET原位成纤共混体系中,分散相PET为球状,均匀分散在连续相PP中;而采用配备有头部直槽混炼件螺杆的单螺杆挤出机挤出制备的PP/PET原位成纤共混体系中,分散相PET为球状和短而粗的微纤,这是因为物料在螺杆的槽中输送前进时,PET先分散成微球,在通过螺杆头部的直槽混炼件时,PET微球受到直槽混炼件过料狭缝的强烈拉伸作用,使部分PET拉伸成短而粗的微纤。
因此,PP/PET原位成纤共混体系中PET的用量、拉伸比和加工设备等对PET微纤的形态都会产生影响,进而影响共混体系的力学性能、流变性能等。
3.3 PP/其他聚合物原位成纤共混体系
PP除了与PA6、PA66、PET等共混制备原位成纤共混体系外,还有研究者利用PTFE、ABS等强度高等特点与PP共混制备原位成纤共混体系,以提高PP材料的结晶和力学等性能。
Zhang等[46]采用2种牌号的PTEF(PTFE-3800和PTFE-1400)在PP中原位成纤,发现PTEF-3800在PP中呈微纤网络状[图16(a)],PP依附于微纤网络结晶[图16(b)],提高了共混体系的力学性能;而PTFE-1400在PP中以液滴状存在,起到异相成核作用,使PP晶粒细化,数量增加,力学性也得到提高。但PTFE微纤比PTFE颗粒更有利于共混体系结晶,微纤抑制晶体生长,促使PP生成微纤网络晶体结构,因而力学性能更加优异。
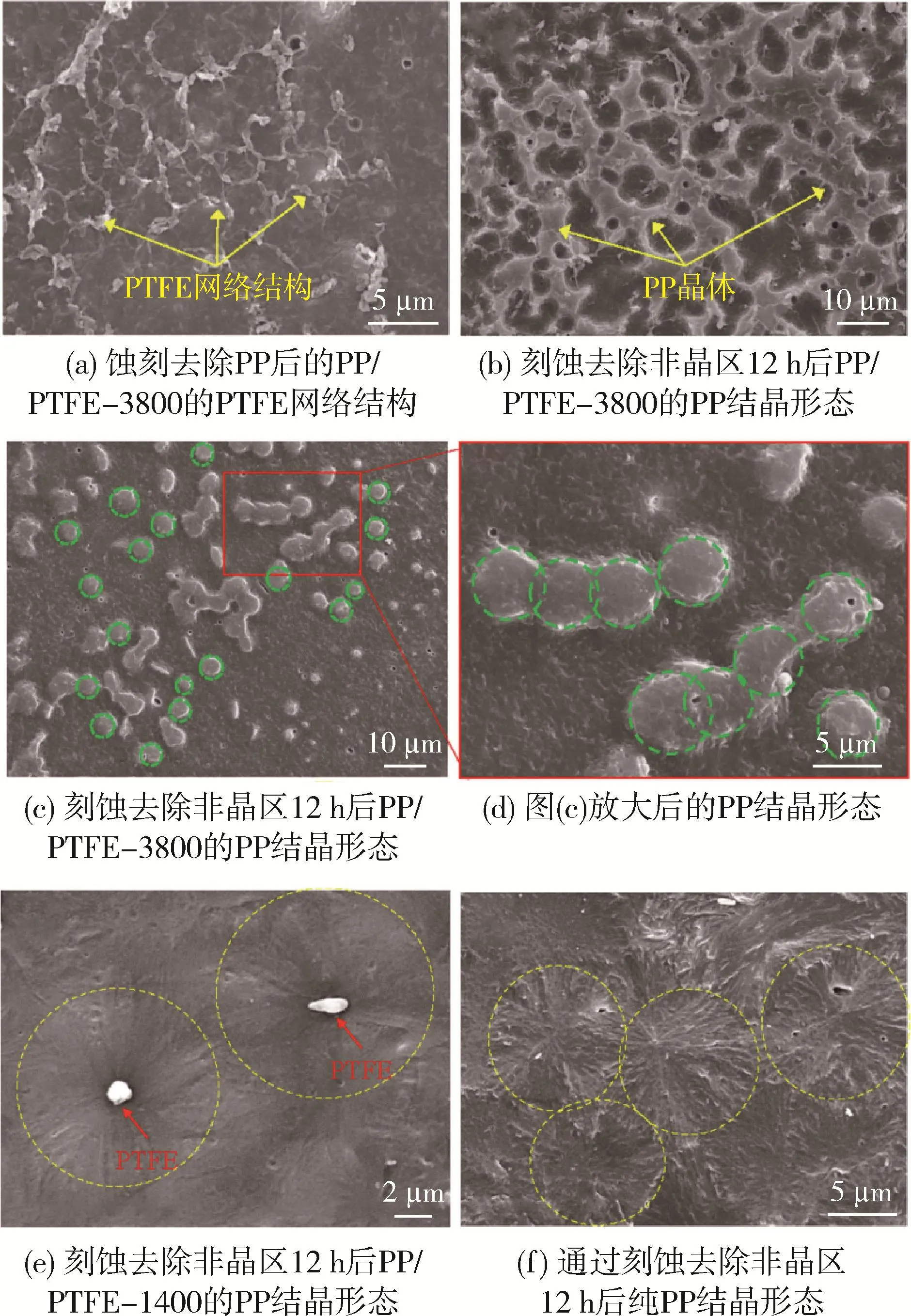
图16 PP和PP/PTFE共混体系的晶体形貌[46]Fig.16 Crystal morphology of PP and PP/PTFE blends[46]
张贻舟[47]采用双螺杆挤出机分别在不同加工温度(190、230℃)和不同螺杆转速(50、200、300r/min)条件下,制备了 PP/ABS(质量比分别为95/5、90/10、85/15、80/20)原位成纤共混体系,发现在ABS含量为10%时,较低螺杆转速(50r/min)与加工温度(190℃)有利于ABS形成纳米级微纤网络结构,微纤平均直径为100 nm;同时采用微型注射机制备了不同加工温度(185和230℃)的原位成纤共混体系,发现加工温度为185℃比加工温度为230℃的共混体系的弯曲强度和弯曲模量分别提高了9.2%和13.7%。
邢栋[48]通过熔融共混挤出—拉伸工艺制备了PP/PPS(质量比为 90/10、85/15、80/20、75/25)原位成纤共混体系,发现随着PPS含量增加,PPS微纤直径增加,PP球晶半径先增大后减小。理论上PPS成纤可增强PP,但PP与PPS界面相容性较差,导致PP/PPS原位成纤体系的拉伸强度比纯PP要低,且随着PPS含量增加而降低,因此需添加相容剂。结果表明,添加相容剂后,随着PPS含量增加,冲击强度逐渐提高,说明相容剂提高了PP与PPS两相界面相容性,形成更多PPS微纤,完善了纤维网络结构。
上述研究结果表明,对于PP/PA原位成纤共混体系,PA的用量、加工温度、相容剂、拉伸作用等对分散相PA在PP中成纤形态影响较大,所形成的PA微纤可在一定程度上改善PP的结晶性能和力学性能等。对于PP/PET原位成纤共混体系,在其中加入相容剂时,更加有利于PET成纤;PET微纤使PP/PET共混体系的力学性能、结晶性能和加工性能等均有一定程度提高。
4 结语
聚合物原位成纤技术问世30多年来,对其研究越来越多,涉及的聚合物共混体系除PP为连续相的体系外,还有以PE等石油基聚合物为连续相的各种共混体系[49-54];现在也有将其用于可生物降解聚合物原位成纤共混体系,如PBAT/PLA[55-56]等。但由于影响共混体系中分散相形成微(纳)纤的因素较多,至今还没有在工业中得到广泛应用。未来还应对各种聚合物原位成纤共混体系中分散相成纤的影响因素进行深入研究,开发出各种稳定的最佳配方体系和工艺参数,使其成为PP等通用塑料高性能化的可行而有效的途径。
