基于全基因组分析技术的鱼类育种技术原理与应用
2022-03-23石米娟张婉婷程莹寅夏晓勤
石米娟 ,张婉婷 ,程莹寅 ,夏晓勤 *
(1.中国科学院水生生物研究所,武汉 430072;2.中国科学院种子创新研究院,北京 100101)
近十余年来,高通量测序技术的快速发展逐步消除了基因组测序的成本与技术壁垒,单个物种基因组的测序组装已由举全球之力[1]变成单个实验室独立承担[2],已完成测序的鱼类基因组数目呈指数上升,“万种鱼基因组计划(Fish 10K)”更是昭示着鱼类后基因组时代的来临[3]。随后,多学科交叉融合以及相应技术的推陈出新则构成了后基因组时代的“主旋律”。全基因组序列与功能注释是所有基因组相关应用的基础,其将序列与功能相联系,极大地提高了各类组学研究以及育种中性状因果位点预测研究的准确性与可读性。此外,对于育种研究而言,全基因组序列信息还为经济性状相关位点的定位与遗传机制的解析打开了方便之门。鱼类很多经济性状是由多个微效基因共同作用而决定的,传统标记密度低且很难定位至基因水平,而全基因组分子标记与表型性状的关联研究更全面、更精准地定位于目标性状相关的基因或分子模块,为育种对象的分子选育和遗传改造提供更为精准的候选靶标。本文梳理了基于全基因组分析的鱼类育种相关技术,介绍了全基因组关联分析(genome-wide association studies,GWAS)、全基因组连锁分析(genome-wide linkage analysis,GWLA)、全基因组选择(genomic selection,GS)等研究进展,旨在推动全基因组分析技术在鱼类育种研究中的应用。
1 全基因组分子标记
1.1 鱼类育种主要分子标记
目前鱼类研究中常用的分子标记有2种:简单重复序列(simple sequence repeat,SSR)标记和单核苷酸多态性(single nucleotide polymorphism,SNP)标记。SSR标记即微卫星(microsatellite)标记,是由几个固定的核苷酸单元组成的一段串联重复序列,其重复单元的数目在不同个体中不一样,因而呈现不同长度的片段,这种差异需要依靠高分辨率的聚丙酰胺凝胶电泳检测[4]。SSR标记的多态性较高,但串联的重复单元在复制时容易发生链滑动,因而SSR在试验和遗传过程中的稳定性都略差。SNP标记是基因组上具有多态性的单核苷酸座位,一般通过测序或芯片杂交进行检测,其稳定性好,但多态性受限于有限的碱基类型(ATCG),实际上绝大多数的SNP标记在群体中只有2种类型,即二态性。简而言之,这2种标记虽然都广泛分布在基因组上,但由于标记的性质不同,两者在多态性、稳定性、检测手段等方面均不相同。
1.2 SSR与SNP标记的应用与局限性
基因组选育分析都需要先获得指定样本中全部候选分子标记的分型矩阵,这一过程在逻辑上包括2个步骤:①候选标记开发;②候选标记分型。对没有参考基因组的物种而言,SSR标记的开发比较耗费资源,而有参考基因组的物种只需要使用程序从基因组序列中进行重复单元的识别与调取即可,已有一些数据库收录并整理了这类标记信息以供使用[5]。SSR标记的分型需要通过大量的PCR扩增和聚丙酰胺凝胶电泳来完成,所需的PCR反应数目为候选标记数和样本个体数的乘积,样本量和候选标记数越大导致试验规模和成本越可观。因此,SSR标记更适用于候选标记数较少或者目标群体较小的研究[6]。
各物种的基因组上SNP含量丰富,其数目一般数十倍甚至百倍于SSR标记。对目标群体的所有选定样本进行测序(基因组重测序或简化基因组测序等)[7-8],然后与参考基因组进行比对,对单一比对位点进行SNP calling等一系列处理[9-10],即可筛选出SNP位点,并且获得它们在所有样本中的分型。这种方法耗费较小,能一次性获得大量的SNP,可以有效弥补SNP多态性低的影响。随着高通量测序技术的普及,SNP标记的应用越来越广,已成为水产育种常用的分子标记类型。
1.3 标记的发掘与改进
参考基因组对于SNP标记的发掘和注释有关键性的作用。虽然缺乏参考基因组也可以依据测序片段的相似性进行聚类而发掘SNP标记[11-13],但这种无参的标记发掘方法极易引入重复序列导致的“伪SNP”位点,而且所发掘的SNP位点没有功能注释,无法进一步解读。随着鱼类基因组信息的逐步完善,有参的标记发掘将成为主流。
高通量测序技术可以有效地降低试验强度,所以也有研究者尝试将SSR标记同高通量测序相结合。但由于SSR序列容易产生“链滑动”,PCR扩增会导致相同的SSR片段最终呈现为不同长度片段的混合物。高通量测序从建库到上机一般需要2次PCR扩增,会将这一问题更加复杂化。如果某些SSR标记在不同个体间的长度差异不大,就容易因测序错误而无法区分,故而高通量测序技术仅适用于SSR标记长度差别较大的样本分型[8,14]。
SSR标记不适合进行高通量测序,SNP标记的分型多态性又太低,已有研究者尝试开发既适用于高通量测序、又有适当分型多态性的标记。微单体型(micro-haplotype,MH)标记是一段包含多个SNP位点的短片段序列,可以通过测序数据获得,多态性较高,已用于草鱼亲子鉴定[15],显示了较好的应用前景。目前,对这类标记的基因组分布情况、突变率和正确性等的研究尚处于开发前期。
2 全基因组标记与表型性状的关联分析
2.1 分析技术
根据计算方式的不同,全基因组大量分子标记与特定表型性状的关联分析可以分为2种类型:①对所有标记逐一进行独立计算分析的单位点关联分析技术;②对所有标记进行整体求解的全位点整体分析技术。
单位点关联分析技术研究单个标记与性状的关联性,从而发掘效应基因或位点,在遗传疾病研究和动植物育种中被广泛应用。从目标性状来看,关联分析可以分为质量性状座位(qualitative trait locus)关联分析和数量性状座位(quantitative trait locus,QTL)关联分析2种类型,大多数经济性状属于数量性状。从研究群体来看,这类研究又可以划分为针对随机群体的全基因组关联分析(GWAS)和针对家系群体的全基因组连锁分析(GWLA)。
数量遗传学研究常基于不同环境和不同家系,对目标表型的方差进行拆分,计算可遗传的基因型值,即育种值,然后分析该值对表型的影响程度,并据此确定个体种质的优劣,这是在育种中被广泛采用的选育和分析技术。早期将一个亲本当作一个整体来研究,仅仅依靠不同代系、不同环境下大量相关个体的表型数据来计算亲本的育种值,后来引入分子标记能极大地增加基因型值计算的准确度。2001年,Meuwissen等[16]又提出了基于随机群体的基因组选择(GS)育种,旨在将群体中所有的QTL都用于估计育种值,而并非依赖人为设定的阈值来界定中高贡献度的QTL座位或其他特定的已知位点。该技术是全位点整体分析技术,在牛[17]、猪[18]的育种中都取得了不俗的成绩。
2.2 GWAS、GWLA和GS的主要区别
GWAS和GWLA都属于单位点关联分析技术,GS则是全位点分析技术。它们都是首先在全基因组范围内建立分子标记与表型性状的关系,然后筛选出选择育种的目标个体或基因操作育种的分子靶标(图1)。GWAS与GWLA的主要差别在于适用群体和关联算法的选择。一般来说,GWAS是在随机群体中利用哈迪温伯格(Hardy-Weinberg,HW)平衡对标记进行过滤,再通过统计检验计算标记和表型相关的概率;GWLA则是利用家系所对应的孟德尔分离比进行标记过滤,并计算待测标记与表型连锁的概率;GS在随机群体和家系中均可计算基因组估计育种值(genomic estimated breeding value,GEBV),从而评估个体基因型对表型的贡献度,只是对于家系数据而言,还需要引入亲缘关系矩阵来计算家系的影响。
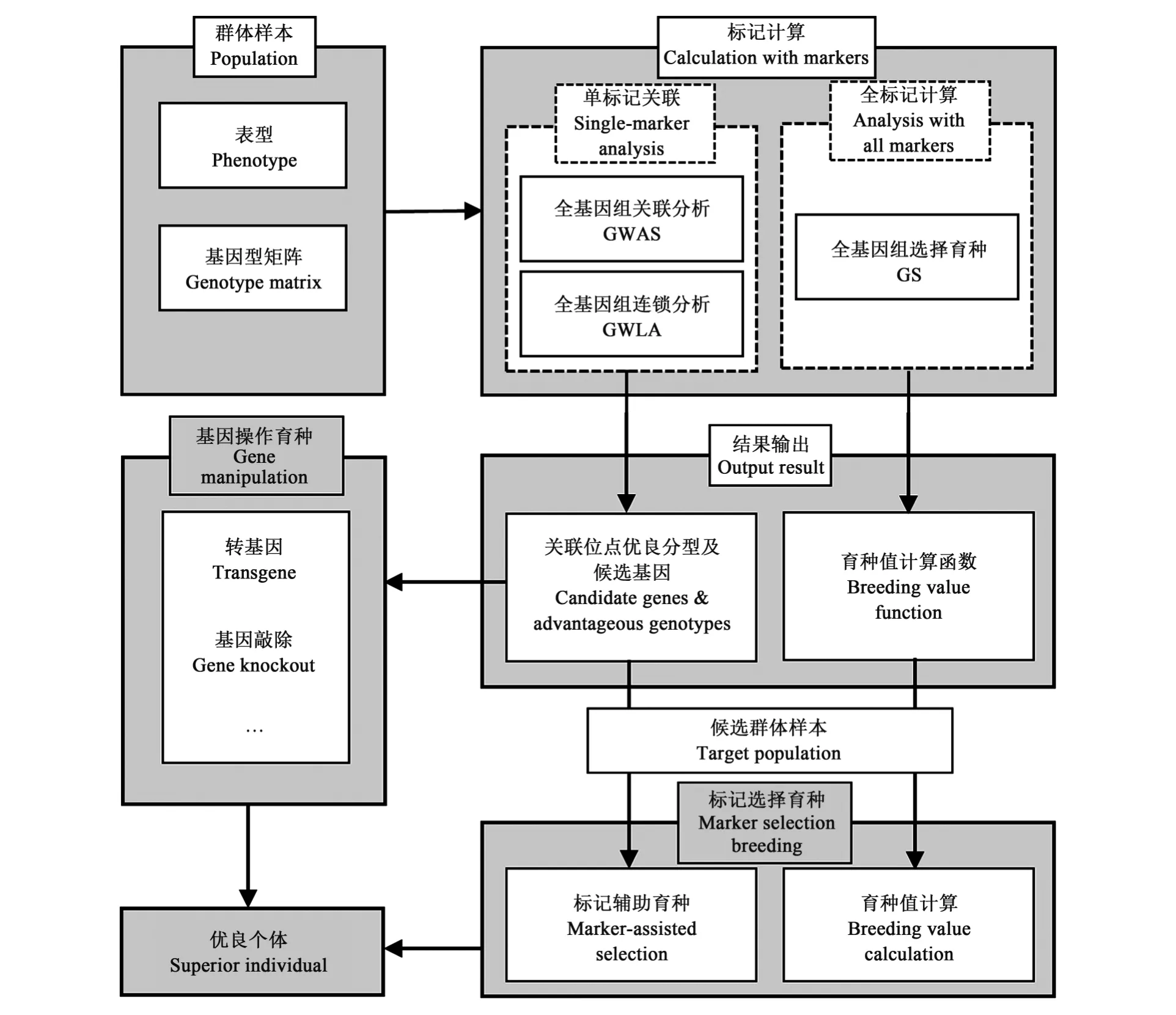
图1 基于全基因组分析的鱼类育种技术Fig.1 Technology of fish breeding based on whole genome analyses
对于单位点关联分析而言,需要根据群体、性状和标记密度的特点选择相适应的算法。这些算法对于性状关联位点的判定大都依据相关统计量的阈值,而这种阈值的设定一般具有主观性。虽然也有一些检测阈值合理性的方法,比如置换检验等,但单位点关联分析往往只适合寻找关联度较高的QTL位点,低贡献度的QTL容易被忽略。而有些数量性状的表型是很多低贡献度QTL位点叠加作用的结果,这无疑影响了该类方法在实际工作中的应用。全位点分析方法虽然不存在这种困扰,但是大量标记的引入导致无法精确求解,只能使用各种近似求解法,而这些方法的结果往往不一致。
3 全基因组标记与表型性状相关联技术
3.1 单位点关联分析技术
单位点关联分析的目的在于检测单个标记是否与目标性状相关联,即基因型的不同是否会导致目标性状的改变。在实际应用中,质量性状与数量性状所适用的关联分析算法不同。对于质量性状而言,根据性状的表型可以将样本分成不同的组别,从而检查不同组别之间的基因型分布特征是否相同。以简单的二元质量性状(如某种疾病的有无)为例,在群体中某个具有3种基因型(AA、Aa和aa)的标记,在对照组(control)中的样本基因型AA:Aa:aa为1:2:1,假设处理组与对照组没有差异,即零假设(H0)。那么,在由40个样本组成的处理组(case)中这3种基因型的期望数目应为10:20:10。若实际观察值为24:12:4,经卡方检验,P=0.006<0.01,说明处理组的观察值极显著地偏离了期望值,零假设不成立,因此可以认为这个标记与处理组的性状相关联。
对于数量性状而言,表型值往往是连续变化的,一般不适合根据表型从群体中区分对照组和处理组,而是比较几种基因型之间的表型数据分布是否存在差异。比较常见的做法是通过某种广义线性模型(generalized linear model,GLM)或混合线性模型(mixed linear model,MLM)来评估包括基因型在内的多种因素对表型的影响。以 MLM 模型(y=μ+αX+βZ+γW+e)为例,个体的表型值(y)被分解成5种成份:①μ为表型的总体均值;②αX表示一些固定效应因子(如群体结构、性别、年龄等)的影响;③βZ为基因型(Z)的遗传标记效应,一般作为固定效应,也有人认为作为随机效应更为恰当[19];④γW一般指个体亲缘关系的影响,是随机效应;⑤e是环境因素带来的随机误差。通过回归分析可得到β的估计值,以该值的大小和统计显著性来判定一个标记是否与某种表型存在直接的关联性。
3.2 全位点整体分析技术
分子遗传学往往关注与性状相关的单个或多个分子位点,而数量遗传学最初是将每个亲本作为一个整体来研究相关个体和表型数据的关系,并通过遗传方差的分解,解析目标性状在代系传递中的规律。作为全位点整体分析技术的主要代表,GS育种延续了这一思路,并以全基因组标记来代替个体样本。GS育种一般需要通过1个训练样本集来估计方程参数,再利用方程计算测试样本集中各个待测样本的育种值,最后根据育种值筛选合适的样本作为优良种质进行培育。
在训练集中,以所有样本的全基因组标记位点为自变量,以样本的目标表型为因变量,用于构建方程。假设有n个标记位点,构建模型为y= ∑aixi+ε,其中,y为表型值,xi代表第i个标记位点,ε为随机误差而ai是需要估计的参数。将训练集中所有样本的表型值以及基因型值带入方程中用来估计ai。这种方法所面临的问题是标记位点数目庞大而检测样本的数目相对较少,即自变量数目远大于因变量数目,自由度不够,造成参数估计因难。针对上述问题常用的解决方法是将SNP效应当作随机效应,再用最小二乘法、GBLUP、BayesA和BayesB以及一些衍生方案,其中以BayesB模型的效果较好[16-20]。
4 全基因组分析技术在鱼类育种中的应用
4.1 GWAS技术在鱼类育种中的应用
4.1.1 GWAS分析过程 GWAS是针对随机群体而设计的关联分析,最初应用于人类疾病相关基因位点的分析[21-23]。经济鱼种类繁多,许多鱼都还处于未经人工选育的阶段,这类鱼的野生群体通常也可以看作一个随机群体。单纯从技术角度来看,GWAS的应用比较简单。在群体中随机选择样本之后,通过测序或基因芯片获得个体的标记分型,然后通过各种统计检验来评估各个标记与表型之间的关联性,包括以下步骤。
①标记预处理。一般会从检出率、最小等位基因频率 (minor allele frequency,MAF)、HW 平衡3个方面对标记进行筛选,过滤不可靠的标记。检出率即检测到该标记的样本比例,较低的标记需过滤掉;MAF较低的标记是测序错误导致的;不符合HW平衡的标记一般需要摒弃。
②群体分层检测。所谓群体分层,是指采用的随机群体样本中包含1个或多个近缘小群体。如果采样群体由几个家系组成,各家系小群体之间的一般性基因组差异很容易被判定为高关联的位点,严重影响GWAS结果的准确性。为了判断群体是否分层,可以对所有样本的二态性标记进行主成分分析(principal component analysis,PCA)[24],观察样本的聚类情况,若样本出现明显的聚类,则表明存在群体分层,需要去除分层样本,或将分层群体属性也作为变量纳入计算。
③位点与性状相关性分析。分析算法的选择与研究的具体情况,尤其是性状类型相关。总的来说,质量性状一般选用卡方检验,计算处理组是否偏离了预期值;而数量性状可选用广义线性模型,检验不同基因型个体的表型值是否遵从相同分布。对于一些特殊的应用场景,比如处理组与对照组数据严重不平衡的情况下,还需要采用其他模型[25]。经过以上计算获得GWAS的原始结果,包括用于评价单个标记与性状相关联与否的P值,一般来说,P值越小,位点与性状的相关性越高。
4.1.2 GWAS的相关问题 由于P值大小受样本的性状、数目及个别样本分型的缺失和错误等的影响,对于GWAS分析的结果还应当尽量通过标记的连锁不平衡(linkage disequilibrium,LD)、标记相关的基因功能注释信息或扩大群体样本量加以验证,才能形成较为可靠的结论。假设有3个SNP位点在所有样本中都呈现紧密连锁状态,即完全的LD。由于技术的原因,其中1个标记在某些样本上的分型可能缺失,而另1个标记在某些样本上分型错误,最后1个标记既无缺失也无错误,这将导致P值本该完全相同的这3个SNP标记最终P值都不相同,甚至差距较大。更糟糕的是,如果P值最小的标记是由于分型错误而产生,再将其作为最终的效应位点会得出错误的结论。
为了解决上述问题,可以在全基因组范围查找状态同源(identical by state,IBS)区域,以代替单个SNP位点,同一IBS区域下的SNP呈完全LD。寻找IBS的简单方法是计算所有标记两两间的LD值(如D’或r2),设定LD阈值来定义高相关的标记群,并将这种标记集合定义为1个IBS。这种方法计算量很大,100万个SNP位点需要计算近5×1011个LD值。因此在实际操作中,一般沿着基因组序列的顺序计算某一区域内标记之间的LD值,然后绘制LD值与标记间距的曲线,确定LD阈值,用于定义IBS区域[26-27]。这种处理方法对基因组的正确性,尤其是SNP位点在基因组上排序的正确性要求较高。所有的上述步骤都有相应的软件可使用,GWAS常用的软件有PLINK[28-29]等。
4.2 GWLA技术在鱼类育种中的应用
4.2.1 GWLA分析过程 GWLA基于家系群体计算标记与性状连锁的概率,一般需要先构建合适的家系群体,获得亲本和大量子代(≥100尾)的全基因组SNP分型矩阵,构建遗传连锁图谱(连锁图),最后基于连锁图来进行QTL定位。
养殖鱼类通常是体外受精且产卵量大,仅靠一对亲本就可以一次性产生大量的后代,能够方便地构建家系群体。然而,其代系周期较长,不易获得纯系,难以像作物那样构建回交(back cross,BC)、F2和RIL等近交系(inbreeding)群体,所以对经济鱼类开展GWLA研究时,往往直接使用两尾杂合亲鱼直接构建子一代家系,即远交系(outbreeding)群体。
从理论上来说,参考基因组是非常精细的物理图谱,可以用于QTL定位。然而,现有大多数鱼类参考基因组的质量达不到GWLA的要求,一些组装错误妨碍了它们的应用,尤其是当SNP的顺序存在问题时,会导致QTL估计出错。相反,连锁图能提供更可靠的标记顺序框架来辅助QTL定位。尽管作物中较为成熟的各类GWLA方法和软件大多基于近交群体,但不能直接用于鱼类的远交系群体,目前也有少数为远交群体设计的连锁图构建软件,较为常用的有JoinMap[30]和OneMap[31-32]。
与GWAS类似,GWLA的QTL定位也是检验表型与单个标记的基因型之间是否存在相关性,常用的统计量是LOD。若仅检测单个标记和性状之间的关联,在有参考基因组的情况下,连锁图存在与否对最终单个标记的P值影响很小,计算P值的统计方法和GWAS也很类似。因此,在许多高密度遗传图谱与QTL定位研究中,如果同时进行QTL定位和GWLA分析,一般能取得很好的一致性[33]。但是在实际过程中,如果分子标记不够密集,则QTL可能被定位在2个遗传距离不紧临的标记的间区。计算这类QTL就需要利用区间作图或复合区间作图[34]等方法获得连锁图。目前常用的QTL定位软件有MapQTL[35]和R/qtl[36-37]。
4.2.2 GWLA的相关问题 鱼类GWLA面临的问题主要为连锁图的构建。连锁相可以辅助计算2个标记的遗传距离,但远交系群体标记间的连锁相是未知的。从分子层面来看,远交系群体全基因组所有的SNP标记可以看作是“假测交”类标记(一个亲本纯合,另一个亲本杂合)和双杂合标记(ab×ab)的混合。未知连锁相对“假测交”类标记影响不大,但对于双杂合的标记而言,只能通过统计推断来判断标记间可能的遗传距离。连锁图的构建就是基于标记间的遗传距离矩阵寻找权重最小路径,以便将所有标记串联起来,这一过程是计算机算法中著名的“邮递员问题”,只能利用各种统计模型或算法进行近似求解[38]。由此可见,远交系的数据需要经过标记距离和标记顺序2步近似求解,严重降低了连锁图的准确性。
如果从一对亲本鱼的全基因组中筛选出所有双纯合的分子标记(aa×bb),其家系可视为近交系群体,就可以使用现有的作物遗传分析软件,基于这些标记在F2代、BC子代等群体中的基因型表现来定位QTL,即在标记层面构建“假近交系”。在该情况下,所筛选标记的连锁相是已知的,常用的作物分析软件或流程皆可使用,但必需放弃大量非纯合的分子标记,甚至是效应位点,这无疑会影响最终结果的准确性和完整性。
4.3 GS技术在鱼类育种中的应用
4.3.1 GS分析过程 GS分析利用全基因组的SNP位点来估计个体的育种值,最早应用于畜牧业中。GS分析首先构建并训练预测方程,然后使用预测方程计算待测样本的育种值。预测方程的训练过程需要获得训练样本的目标表型数据以及全基因组的SNP位点分型矩阵。在后期获得测试样本的目标表型值之后,也可将待测样本再纳入训练集进行预测方程的再优化,为后续的鱼种选育提供更精准的预测。该步骤是GS育种的关键,好的预测方程能在测试样本中获得更为精准的育种值,而育种值正是选择育种的依据,据此可以选择合适的样本作为亲本进行培育。
4.3.2 GS的相关问题 训练样本集可以来源于随机群体或家系群体。选择随机群体时,对GWAS产生负面影响的因素也同样会降低GS算法的准确性,比如群体分层和标记的分型错误或缺失,应在计算模型中纳入亲缘关系矩阵和利用多SNP的IBS来降低相应的负面影响[39]。相比之下,家系群体更具优越性,在家系中育种值计算的准确度远高于随机群体[40-41],尽管家系群体性状表型的丰富程度一般低于随机群体。无论哪种群体,育种值的准确度都会随着目标性状遗传力的降低而降低,而随着训练集样本数目的增多而上升[40-41]。总的来说,具有真实效应的QTL位点在所使用的全部位点中的占比以及这些效应位点在训练集和测试集样本中分型的准确性决定了GS分析结果的准确度。因此,研究人员尝试通过GWAS来尽可能的寻找效应QTL位点,减少噪音自变量,从而提高GS的准确度[42-43]。然而这样容易漏掉许多低效力的QTL位点,与设计GS的初衷相悖。
鱼类的GS研究一般使用随机群体[43-44],可用软件较多,如 ASREML(http://www.vsni.co.uk/software/asreml)或 DMU(http://dmu.ghpc.au.dk/dmu/),鱼类育种研究者也在探索更为精准的全基因组育种值估计方法[44-45]。对于鱼类家系,尤其是远交家系,目前尚无相应的GS分析软件和流程。
5 全基因组育种技术存在的问题和建议
单位点关联分析与整体关联技术本质上并不存在群体选择的差异性。虽然水产育种研究中GWLA使用较多[46],但技术的选择完全取决于研究目的。若是为了获得候选的基因及其单体型,单位点关联分析技术是首选,所获得的基因和单体型可以结合分子标记辅助选择(molecularmarker assisted selection,MAS)育种或者基因编辑技术来加快育种进程;如果需要从大量的子代群体中寻找合适的后备亲本,则GS更为恰当。
需要注意的是,从目前的实践来看,无论是关联分析找到的SNP位点还是GS构建的育种值计算方程,都仅适用于相近的群体,即训练集来源于待测试集,或者两者间存在紧密关系。如果跨群体使用,效果都欠佳,这一点GS表现尤其明显,这可能是由于在分析过程中都仅考虑了可以稳定遗传的加性效应,而没有将上位效应等其他基因互作纳入计算。现实中生物体基因的互作复杂程度远超现有研究,如何解决这类问题也是遗传研究者们的研究方向之一。
6 结语
颠覆性的技术必然会带来相关行业与相关技术的革新,高通量测序技术的普及为生物学各研究领域都注入了新的活力。随着各种经济鱼类基因组的测序完成,鱼类育种技术也在悄然发生变化,鱼创新品种选育工作正在跨入一个新的时代。比如与高通量测序相结合的液相芯片技术和多重PCR技术,可以一次性实现对大量样本多个靶向位点的测序,直接通过软件分析获得结果,而不需要人工解读电泳图,因此速度快、效率高,极大地节省了人力、物力资源。在靶向位点已经确定的情况下,这一技术可广泛地应用于物种遗传多样性检测、品种/品系鉴定、家系与亲缘关系分析乃至分子标记辅助育种,尤其适用于成千上万甚至更高数量级样本的高通量检测。毫无疑问,高通量测序将使得各种鱼类不同性状的分子标记或QTL鉴定越来越普遍。随着各类分子标记与位点的逐步发掘,收录这类分子信息的鱼类育种数据库也将出现,将分散的零碎信息集中起来。这类信息平台的充实和完善将进一步方便全基因组水平的遗传分析,使研究人员有可能为特定性状筛选出强相关的分子模块或标记,以少量标记实现对候选亲鱼的大规模筛查,从而大大缩短育种周期,加快经济鱼类的育种进程。在这个过程中,如何借鉴其他物种经验尽快开发出真正适合鱼类特点的整套基因组育种分析技术,同时搭建和维护各种经济鱼类的分子信息综合平台,值得鱼类遗传学研究者思考和努力。
