细胞自噬在恶性骨肿瘤中的研究进展
2022-03-23对红白锐
对红 白锐
细胞自噬 ( autophagy ) 是指通过溶酶体分解细胞内成分的过程。而且细胞自噬通过降解胞内成分和向细胞提供降解产物,在维持和调节细胞动态平衡方面起着重要作用。自噬广泛存在于真核细胞的病理生理过程中,随着对自噬研究的不断深入,其在肿瘤中的作用日渐引起广泛关注。骨和软组织肉瘤是一种间充质来源的肿瘤,虽然仅占所有人类癌症的不到 1%,但恶性程度较高,易致残。尽管晚期化疗和手术对其有一定改善,但大多数恶性骨肿瘤的治疗方式在过去数十年间仍然停滞不前,导致患者饱受疾病的折磨,精神压力、经济压力、甚至是摧毁一个个家庭,因此探索恶性骨肿瘤的发病机制是迫在眉睫的。已知常见的恶性骨肿瘤包括骨肉瘤 ( osteosarcoma,OS )、尤文氏肉瘤 ( ewing sarcoma,ES )、未分化高级别多形性肉瘤 ( undifferentiated high-grade pleomorphic sarcoma,UHPS )、脊索瘤 ( Chordoma )、恶性骨巨细胞瘤、浆细胞性骨髓瘤等,确诊时往往多伴远处转移且 5年生存率较低。目前用于其早期检测的肿瘤标志物和治疗手段十分有限,为突破这一瓶颈,近年来医学研究人员正努力探索细胞自噬在恶性骨肿瘤中发挥的作用。
现从信号传导通路、肿瘤治疗中的应用等方面简要探讨各类常见恶性骨肿瘤中细胞发生自噬的相关机制,为其临床诊断和治疗靶点提供参考文献。
一、细胞自噬
细胞自噬最早于 1962年用电子显微镜在人肝细胞中观察到的一种溶酶体降解途径,是细胞通过双层膜包裹待降解物形成自噬体 ( autophagosome ),然后运送到溶酶体形成自噬溶酶体,降解其所包裹的内容物,以实现细胞本身的代谢需要和细胞器的更新,因为所有过程均在同一细胞中完成,所以被称为自噬。它将受损或过剩的细胞成分降解为基本的生物分子,然后再循环回胞质,通常被认为是在应激或营养缺乏等应激条件下保护细胞的一种促生存机制。
细胞自噬发生的过程大致分为四个阶段:( 1 ) 起始:胞质中首先形成双层分隔膜,随后向两边延伸成自噬泡;( 2 ) 延伸:自噬泡将需降解的胞质物质收入泡中,然后封口,称为自噬体;( 3 ) 成熟:自噬体与内涵体融合,形成自噬内涵体,或与溶酶体结合形成自噬溶酶体;( 4 ) 降解:自噬溶酶体内有许多酶,实现底物的降解。
到目前为止在哺乳动物中有三种不同类型的自噬:巨自噬、微自噬和伴侣介导的自噬。巨自噬是最主要的自噬途径。有文献报道,巨自噬发生机制如下:在细胞应激时,mTOR 被灭活,这使 ULK 复合体进一步被激活。ULK复合体的激活包括 ULK1 依赖的 Atg13、FIP200 和 ULK1本身的磷酸化。这些磷酸化过程是自噬启动所必须的。ULK1 进一步磷酸化 Ambra1,后者与微管上的 PIK3C3 复合物相互作用,形成的 Ambra1-PIK3C3 复合物从微管中释放并转运到自噬小体形成的主要部位 —— 内质网上,在自噬小体成核过程中,PIK3C3 复合物产生 PI3P,与DFCP1、Atg2 和 WIPI1 相互作用,并招募其它参与膜伸长的 Atg 蛋白。Atg7 和 Atg10 介导 Atg5-Atg12 复合物的形成,Atg5-Atg12 复合物随后与 Atg16L1 结合,生成 Atg16L1复合物。LC3 被半胱氨酸蛋白酶 Atg4 裂解成 LC3-Ⅰ。LC3-Ⅰ 由 Atg7、Atg10 和 Atg16L1 复合物介导,与磷脂酰乙醇胺 ( PE ) 结合生成 LC3-Ⅱ。自噬体形成后,自噬体外膜与溶酶体膜融合,将自噬体内容物释放到溶酶体腔内,自噬体内膜被降解,产生的氨基酸 ( AA )、游离脂肪酸( FFA ) 等被释放回胞质中( 图 1 )。还有一种参与自噬调节的相关因子是 Beclin-1。Beclin-1 是 Beclin-1 VPS34 Ⅲ类磷脂酰肌醇 3 激酶复合物活性的重要决定因素,在自噬成核过程中 ULK1 复合物磷酸化并激活 Beclin-1-Vps34 复合物。Beclin-1 / Vps34 复合体是一个动态的多蛋白组装,在巨自噬过程中是必不可少的。Beclin-1 / Vps34 核心复合物的活性受 Beclin-1 结合蛋白的调节。而 Bellin-1 通过进化保守结构域 ( ECD ) 与 Vps34 相互作用,促进自噬。Beclin-1 和 Vps34 的结合受 Beclin-1 和 Vps34 翻译后修饰的调节。
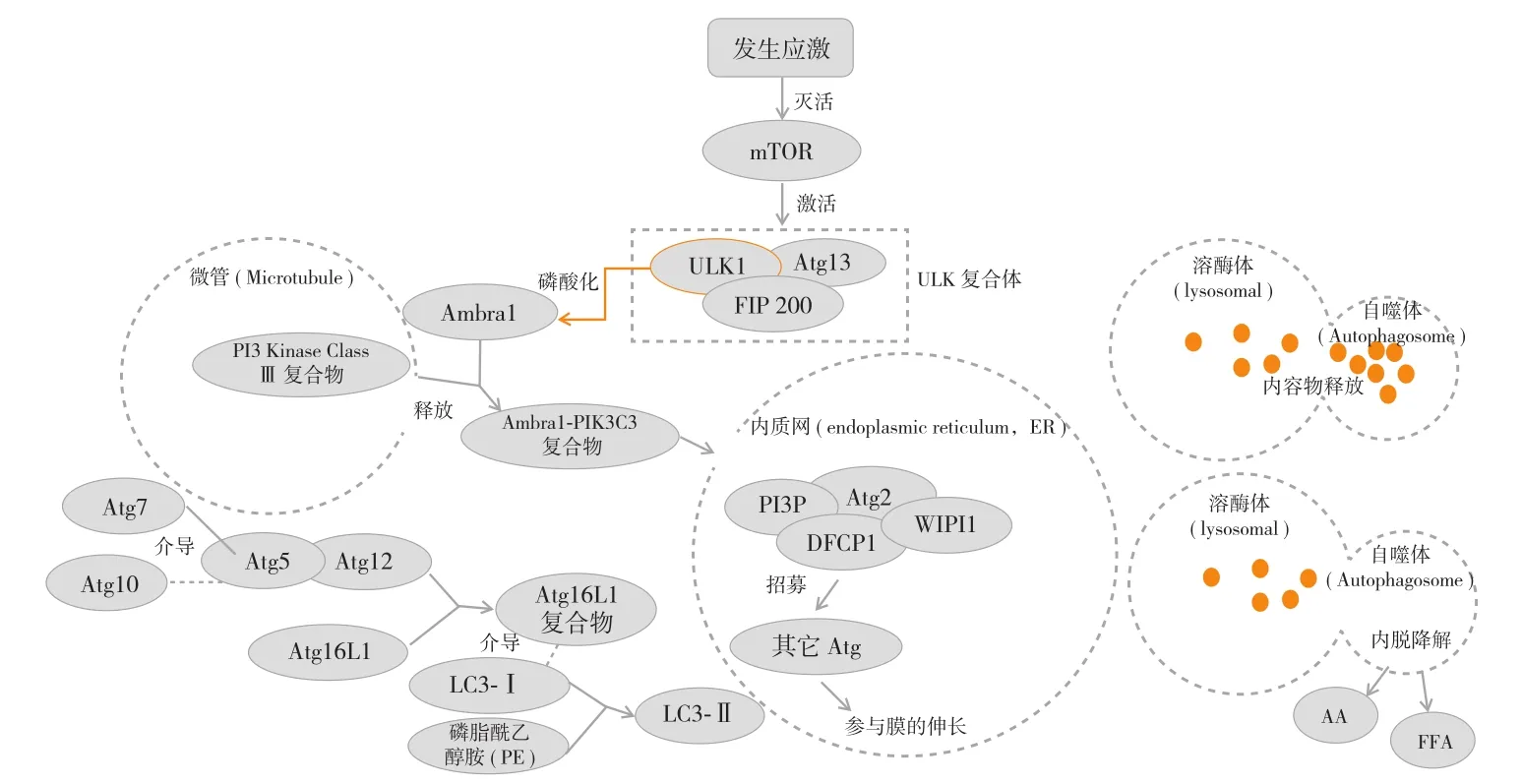
图1 巨自噬机制图 ( 注:实线代表信号传导过程 虚线代表细胞器或复合体 )Fig.1 Mechanisms of macrophage autophagy ( Notice: The solid line represented the signal transduction process and the dotted line represented the organelle or complex )
二、细胞自噬与恶性骨肿瘤
根据 WHO 骨软组织肿瘤分类指南以及生物学潜能的不同将骨肿瘤分为良性、中间型 ( 局部侵袭性或偶见转移型 ) 和恶性三个级别,其中属于恶性级别的骨肉瘤、ES、UHPS 及脊索瘤等恶性骨肿瘤存在患者确诊时已有远处转移、5年生存率较低、复发率高、预后较差等一系列治疗难题,故本综述重点讨论上述四种常见恶性骨肿瘤中自噬机制,从而探索治疗恶性骨肿瘤的新方向。骨肉瘤、ES、UHPS 及脊索瘤的自噬相关机制研究相对较多,其余恶性骨肿瘤中发生自噬机制的文献极少。对上述几种恶性骨肿瘤中自噬发生的相关机制逐一阐述如下。
1. 骨肉瘤的自噬发生机制:骨肉瘤是最常见的原发性恶性骨肿瘤,主要发生在儿童和青少年,好发部位包括股骨远端 ( 43% )、胫骨近端 ( 23% ) 和肱骨 ( 10% ),对于确诊时已经发生转移或复发的患者,5年生存率低于 30%。近几年来对于骨肉瘤由多种药物强化治疗和手术切除联合治疗的方案已被证明具有显著的药物毒性以及高复发率,人们对新治疗策略极为迫切。骨肉瘤的细胞自噬的发生机制大致如下。
( 1 ) 由上游介质激活 Beclin-1 复合体诱导细胞自噬:Beclin-1 相关的自噬相关关键调控因子 ( Barkor ) 是一种特异性自噬蛋白,在人骨肉瘤细胞自噬诱导中起关键作用。在饥饿条件下,敲除 Barkor 基因的人骨肉瘤细胞自噬能力下降,表现为 LC3-Ⅱ 和自噬小体形成减少。Barkor 的重新表达增加了自噬。在此证明 Barkor 直接与 Beclin-1 相互作用,并与抗紫外线辐射基因产物 ( UVRAG ) 竞争,UVRAG 是一个与 Vps34 复合体结合的额外基因,是自噬的启动子进而可以诱导自噬。
( 2 ) 视网膜母细胞瘤 ( Rb ) 基因诱导细胞自噬:Rb基因通过抑制 E2 转录因子 1 ( E2F1 ) 激活骨肉瘤中的自噬,而 E2F1 又下调 Bcl-2,从而激活 Beclin-1 / Vps34 复合体。有研究表明,Rb 导入 SAOS-2 细胞后,通过上调Beclin-1 的表达诱导自噬。过程中也没有观察到 mTOR 磷酸化的变化这也间接说明 Rb 诱导的自噬和 E2F1 介导的Bcl-2 表达比起 mTOR 途径与发生自噬更相关。
( 3 ) 高迁移率族蛋白 1 ( HMGB1 ) 诱导细胞自噬:HMGB1 是一种染色质结合核蛋白,通过控制 Beclin-1 /Vps34 复合物的形成和降低骨肉瘤对化疗的敏感性来诱导自噬。
( 4 ) STAT3 诱导细胞自噬:STAT3 通过抑制蛋白激酶R ( PKR ) 来抑制人骨肉瘤细胞的自噬,PKR 进而抑制真核细胞起始因子 2α ( eIF2α ) 的磷酸化,这也正是激活自噬所需的重要步骤之一,从而进一步影响自噬的发生。
2. ES 的自噬发生机制:1921年,詹姆斯·尤文首次描述了 ES,它主要发生在骨盆、下肢长骨骨干和胸骨。ES 是在儿童和成年人中仅次于骨肉瘤,主要累及的人群是儿童和成人,发病高峰为 15 岁。约有 25% 的患者有远处转移,最常见的转移部位是肺,其次是骨和骨髓。最近的研究表明 ES 中可能发生以下几种自噬机制。
( 1 ) TRIM 诱导细胞自噬:TRIM 蛋白的特征结构包括一个环 ( Ring ) 结构域 R、一个或两个盒 ( B-box ) 结构域 B、位于 N-末端的卷曲 ( Coiled coil ) 结构域 CC 和 C-末端具有可变结构的结构域。研究过程中,进一步发现TRIM3 的一个新功能,即 TRIM3 可以作为 Beclin-1 的 E3泛素连接酶进而影响细胞自噬的发生发展。还发现 TRIM3在 ES 组织中的表达增加,并被 EWS-FLI1 上调。TRIM3 还能抑制 ES 细胞的自噬,并直接与 Beclin-1 相互作用,促进Beclin-1 蛋白酶体的降解,从而抑制 ES 细胞的自噬。
( 2 ) 2-甲氧基雌二醇 ( 2-ME ) 诱导细胞自噬:Lorin等发现抗肿瘤化合物 2-ME 以 p53 依赖的方式诱导自噬,JNK 激活受 p53 调控,强调了 p53、自噬、JNK 激活之间的联系,他们还发现 2-ME 通过激活 JNK 上调 ES 细胞中损伤调节的自噬调节器 ( DRAM )。DRAM 是一种 p53靶基因,下调 DRAM 的表达可以减少 2-ME 诱导 ES 发生自噬。还有研究发现在饥饿条件下,JNK 的激活导致Bcl-2 的磷酸化,通过破坏 Bcl-2 和 Beclin-1 之间的相互作用来增强自噬。
( 3 ) ES 断裂点区 1 ( EWSR1 ) 和 FLI1 ( Friend 白血病病毒整合位点 1 ) 通过上调 ATG4B 诱导自噬:85% 的ES 患者是 t ( 11;22 ) ( q24;q12 ) 染色体易位,从而导致 EWSR1 和 FLI1 基因的融合,形成 EWS-FLI1 病理融合基因。EWS-FLI1 是 ES 中最常见的融合蛋白,它与本综述介绍的细胞自噬相关。在哺乳动物中有四种不同的 ATG4 类似物表达 ( ATG4A、ATG4B、ATG4C 和ATG4D ),其中 ATG4B 对自噬过程至关重要,对不同形式的 LC3 和同源物具有最广泛的底物谱。最近有文献报道,在 ES 细胞中过表达 EWS-FLI1 可以促进自噬,通过自噬的两个重要标志物 p62 ( SQSTM1 ) 的减少和 LC3B-Ⅱ的增加来表现。EWS-FLI1 过表达显著上调 ATG4B,ATG4B 也被证明调节自噬。EWS-FLI1 诱导了 ATG4B 的表达,并且观察到 EWS-FLI1 与 ATG4B 启动子相互作用,这表明 ATG4B 是 EWS-FLI1 的转录靶点。此外,ATG4B 的过表达显著促进了 ES 细胞的自噬,而沉默 ATG4B 则显著抑制了自噬。
( 4 ) microRNA ( miRNA ) 诱导自噬:虽然前期研究已经证明 ES 中的自噬发生机制有很多种途径,但目前公认的自噬机制为 microRNA ( miRNA ) 在不同类型的癌症中调节自噬的下游生物学行为,如启动、成核、延长和完成。有文献报道,miR125a 和 miR351 靶向破坏 UVRAG mRNA。众所周知,抗紫外线辐射相关基因 ( UVRAG )可能通过与 Beclin-1 相互作用来调节自噬,从而影响细胞自噬。
3. UHPS 的自噬发生机制:又称恶性纤维组织细胞瘤( malignant fibrous histiocytoma,MFH ),在最新的 WHO 骨与软组织分类中命名为 UHPS,在软组织肉瘤仅占成人恶性肿瘤的 1%,其中 UHPS 是成人常见的软组织肉瘤之一,1963年,Ozzello 等首次提出。UHPS 中发生细胞自噬和细胞凋亡之间存在着错综复杂的关系,最近的研究表明,在接受抗肿瘤药物治疗的细胞中,自噬以一种自卫机制发挥作用,而阻断自噬可以触发凋亡的激活。UHPS 发生自噬多与 mTOR 蛋白相关。mTOR 蛋白是一种重要的丝氨酸 / 苏氨酸激酶,属于磷脂酰肌醇-3-羟基激酶 ( PI3K ) 相关激酶家族。Nakamura 等研究中得出结论,mTOR 抑制剂替西罗莫司对 Nara-H 细胞 ( UHPS 细胞株 ) 的增殖有抑制作用,并降低了 mTOR 通路相关蛋白4E-BP1、p70S6K 的磷酸化水平,另一方面,促进 Atg12-ATG5 复合体和 LC3-Ⅱ 的表达,诱导 Nara-H 细胞自噬。
王少云等通过在 UHPS 患者手术切除组织中进行免疫组化染色发现,UHPS 组织中 Beclin1、LC-3 高表达,发生机制猜测为 UHPS 细胞恶性程度高,生长迅速,肿瘤中央区发生缺血缺氧,肿瘤细胞通过自噬为其生存提供所必须的成分,进而保证肿瘤细胞的存活及肿瘤的进展,而抑制 UHPS 细胞自噬或许可以抑制肿瘤细胞的生长,这也与上述抑制自噬的发生对 UHPS 的治疗有很大的帮助相吻合。
4. 脊索瘤中与自噬相关机制:脊索瘤是一种起源于脊索残余组织的少见原发恶性骨肿瘤,好发部位为骶骨和颅底部。多数骶骨脊索瘤患者症状较隐匿,就诊时肿瘤往往体积较大,手术切除困难,围术期风险大,术后复发率高。它对化疗高度不敏感,而且缺乏有效的治疗方法。因此,需要研究新的治疗方法。然而,脊索瘤细胞如何产生细胞自噬的机制尚不明确,仅有少量文献报道自噬参与的其它机制,有学者发现,自噬反应可以支持癌细胞在不利的微环境条件下 ( 如化疗 ) 的存活、增殖和生长,从而发生耐药。Wang 等研究证明化疗可以促进脊索瘤细胞的自噬活性,还发现通过 UPR 的PERK / eIF 2α 臂显著加重内质网应激,敲除 KRT 8 可阻断晚期自噬,使脊索瘤化疗增敏。还有研究证明,PAS 糖原染色实验发现脊髓瘤细胞株 MUG-Chor1 和 U-CH2 中含有大量糖原,ELMI 分析显示在自噬细胞、自噬小体和空泡中存在糖原,这表明脊索瘤细胞通过自噬“收获”了细胞内的糖原储存。这也更加证明了细胞自噬可以为肿瘤细胞在不利环境下提供能量,从而使肿瘤细胞存活、增殖和生长。因此阻止细胞自噬的发生或许可以成为治疗脊索瘤的新方向。
三、细胞自噬在恶性骨肿瘤治疗中的应用
恶性骨肿瘤不仅侵袭性高,而且易发生远处转移。治疗方法多为手术切除、化学治疗、放射治疗。虽然一些中药可以用来减少毒副作用,增强放化疗的疗效,但治疗效果往往达不到患者、家属以及骨科医师满意。越来越多的研究表明,自噬在肿瘤细胞中的功能和作用在不同状态下有不同的表现:抑制或促进肿瘤的发生发展。这给科研工作者们起到了良好的启发作用,例如可以抑制或促进细胞发生自噬,提高肿瘤对药物的敏感性来改善恶性程度较高肿瘤的治疗效果,这也提示着细胞自噬可作为治疗的潜在手段之一。
近几年,已经开发出一些具有抑制自噬药物,从作用机制上分两类:早期为阻断自噬小体形成的 3-甲基腺嘌呤( 3-MA )、LY294002 等;晚期抑制自噬小体溶酶体融合和降解的氯喹 ( CQ )、羟基氯喹 ( HCQ ),CQ 和 HCQ 能够抑制细胞保护性自噬,导致癌细胞对不同化疗药物的敏感性不同等。还有一些研究证明,核苷类似物吉西他滨可以通过降低 Akt 和 mTOR 的磷酸化和 Beclin-1 / Vps34 复合物的激活来诱导骨肉瘤的自噬。其它研究证实,一些化疗药物,如顺铂和组蛋白去乙酰化酶 ( HDAC ) 抑制剂也可以通过增加线粒体中 ROS 的产生来诱导自噬。
细胞自噬也可以与其它生物学行为共同影响肿瘤的发生发展,例如凋亡。Nakamura 等发现联合应用 mTOR抑制剂替西罗莫司和自噬抑制剂用 3-MA 可抑制自噬并诱导细胞凋亡。其中联合应用可以使 MFH 细胞减少自噬的同时发生凋亡,从而达到有效治疗 MFH 的目的。
总之,目前原发性恶性骨肿瘤的治疗仍然依赖于手术、化疗、放疗和靶向药物治疗,随着化疗及靶向治疗药物的耐药性及不良反应的出现,对于晚期恶性骨肿瘤及复发患者的治疗仍然处于瓶颈期。细胞自噬在恶性骨肿瘤的增殖、侵袭、转移过程中发挥着重要作用,其作用机制极其复杂。虽然有文献报道了部分恶性骨肿瘤中的自噬机制,如骨肉瘤、ES、UHPS、脊索瘤等,但仍有一些恶性骨肿瘤如恶性骨巨细胞瘤、浆细胞性骨髓瘤等的自噬发生机制报道极少,具体机制仍不明确,生物学作用及意义仍需进一步研究。所以,探索自噬相关信号传导通路、解析了分子间的相互作用以及明确细胞自噬发生机制,探索细胞自噬是否具有肿瘤发生的早期监测作用是极为重要的,这或许可为恶性骨肿瘤的临床治疗提供新的研究方向,为研发新靶点药物提供理论依据,为人类攻克癌症提供新的突破。
