基因表达对膀胱癌生存结局的预测:稀疏与混合Cox模型的实证比较研究*
2022-03-17徐州医科大学公共卫生学院流行病与卫生统计学系221004陆皓杰黄水平
徐州医科大学公共卫生学院流行病与卫生统计学系(221004) 陆皓杰 曾 平 黄水平
【提 要】 目的 研究稀疏Cox(coxlasso)与混合Cox模型(coxlmm)在全基因表达数据中对膀胱癌预后的预测表现。方法 通过计算一致性指数(C-index)评价两种模型在膀胱癌全基因表达数据中(TCGA,GSE31684和GSE13507)的预测精度,同时在混合Cox模型中将膀胱癌的生存方差划分为临床(PCE)和转录组(PGE)两部分。结果 当TCGA数据集为训练集时,coxlmm预测能力(C-index=0.676)高于coxlasso(C-index=0.655),两者在外部验证集的C-index分别为0.527和0.534。当三个合并数据集为训练集时coxlmm(C-index=0.671)比coxlasso(C-index=0.650)的预测精度提高2.1%。当GSE31684为训练集时,coxlmm(C-index=0.553)比coxlasso(C-index=0.550)的预测精度提高0.3%,两个模型在外部验证集上的C-index分别为0.632和0.633。生存方差划分表明膀胱癌的临床贡献高于转录组的贡献(PCE=14.95%,PGE=10.88%)。结论 成功构建了一种用于膀胱癌的预后预测的coxlmm模型,揭示了整合全转录组信息可在一定程度上提高膀胱癌预后预测能力。
膀胱癌(bladder cancer)是最常见的癌症之一,全世界每年新发病例约54.9万,死亡病例约20万,其中男性的发病率和死亡率均高于女性,分别为9.6/10万和3.2/10万[1]。根据其肿瘤分期,膀胱癌可以分为肌肉浸润性膀胱癌(MIBC)和非肌肉浸润性膀胱癌(NMIBC),其中MIBC约占膀胱癌早期诊断病例的25%,NMIBC进展为MIBC的比例高达10%~15%[2-3]。随着医疗水平的不断提高,膀胱癌的预后治疗也取得了长足进步;然而,由于膀胱癌疾病高度异质的缘故[4],MIBC患者的预后仍然较差。不同的遗传标记为深入研究疾病的遗传基础、发展新的诊断技术和治疗方法提供了全新的视角[5],因此整合大规模组学数据同时联合临床信息为膀胱癌预后评价带来了新的工具[6]。
在先前的研究中组学信息已经被广泛应用于膀胱癌的预后预测。然而,先前的研究只是将临床信息和个别生物标志物纳入预测模型;例如,有研究者仅将4个基因(TMPRSS11E,SCEL,KRT78和TMEM185A)纳入膀胱癌预后预测模型[7]。另一些研究者通过降低数据维度的方法(如Lasso方法)提取重要生物标志物进行预后预测[8]。从统计方法角度看,这些方法可被认为是稀疏模型(sparse models),因为其明确假设只有少部分组学信息对预测有用。虽然稀疏模型已经被证实能够提高患者预后预测的准确性,也利于临床实践应用;然而,单个或少数生物标志物的预测性能往往不稳定,而生物标志物的组合能提高预测性能[9]。最近的研究表明,将100、300和5000个mRNA整合到模型中时,预测精度可从0.58、0.62提高到0.64[10],表明在一定程度上纳入更多生物标志物可以进一步提高预测效果。
在遗传预测方面,混合模型(mixed models)同样也展现出较高的准确性[11-13];与稀疏模型不同,混合模型假设所有基因都参与了疾病进展并且对疾病均有影响。很显然,混合模型和稀疏模型的预测效果优劣取决于上述假设与真实情况是否吻合。然而,实际中基因与疾病的真实关系往往未知;因此,对混合模型和稀疏模型的预测性能进行比较和评价具有重要的意义。有研究者利用全基因组表达数据对比评价了九种遗传预测模型方法(包括稀疏模型和多基因模型)[14]。本文通过整合公开的膀胱癌数据,使用C-index来比较混合Cox模型(coxlmm)与稀疏Cox模型(coxlasso)对膀胱癌的预后预测表现;同时在混合Cox模型中将膀胱癌的生存方差划分为临床和转录组两部分。
材料和方法
1.研究人群
从癌症基因组图谱(TCGA)[15]下载了430名膀胱癌患者的信息,保留同时具有基因表达数据和临床信息的345名患者,同时将年龄、性别、肿瘤分期和淋巴结状态作为临床协变量纳入研究。其中男性254人(73.6%),女性91人(26.4%),平均年龄为68.2岁(34~89岁)。从美国国家生物技术信息中心(National Center of Biotechnology Information,NCBI)的基因表达综合数据库(gene expression omnibus,GEO)中下载基因表达谱膀胱癌样本数据集GSE31684和GSE13507[16-17]。GSE31684数据集中男性有58人(75.3%),女性有19人(24.7%),平均年龄为67.0岁(42~85岁);在最后随访中有32名患者死于膀胱癌,有22名患者死于其他原因,其余23名患者仍然存活。GSE13507数据集中男性有134人(81.7%),女性有30人(18.3%),平均年龄为65.2岁(24~88岁);在最后随访中有68人死于膀胱癌。这三个数据集的各项临床特征和生存信息详见表1。
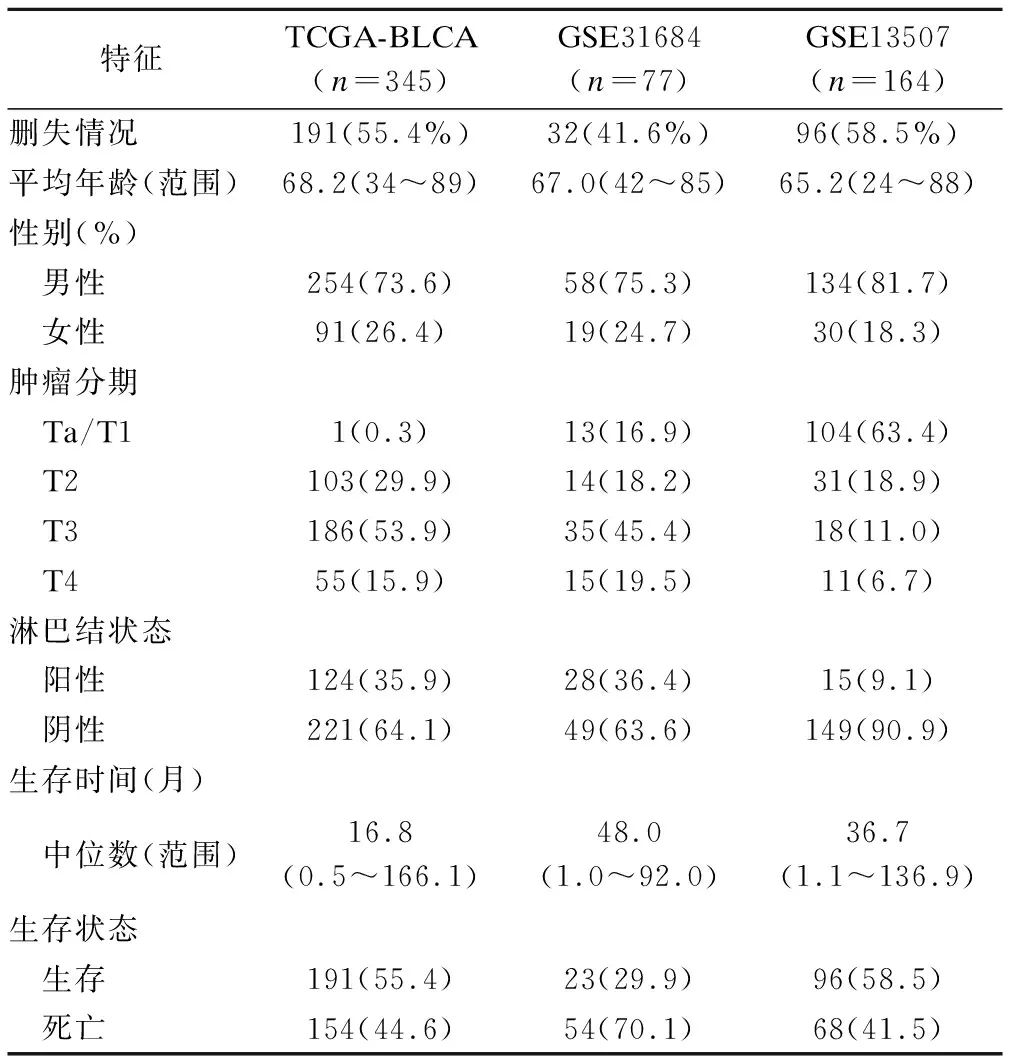
表1 膀胱癌患者的临床和生存信息
2.基因表达的数据集
我们将原始基因表达的count数转换为log2(count+1)并通过R包limma中的normalizeQuantiles函数进行正规化处理[18-19],剔除没有名称和表达量为0高于50%的基因之后还剩下29668个基因。使用R包GEOquery[20]下载GSE31684基因表达,该数据已经过GCRMA算法归一化处理[21];从R包hgu133plus2.db中的hgu133plus2SYMBOL获得探针与基因之间的对应关系。GSE13507基因表达数据经过分位数标准化和log2转换[17],从R包illuminaHumanv2.db中的illuminaHumanv2SYMBOL获得探针与基因的对应关系。剔除两个数据集中没有对应基因名称的探针,当同一基因对应多个探针时,将表达量平均值最高的探针作为基因对应的唯一探针,然后把这组中的其他探针剔除,最后删除表达量为0超过50%的基因之后两个数据集分别保留了20174和15685个基因。将三个数据集的基因表达数据按基因名合并后,保留12517个共同的基因进行后续分析。此外,使用R包ComBat[22]中的sva消除三个数据集的批次效应[23]。
3.一般Cox模型
设Xi为个体i的临床协变量(如TCGA数据集中的肿瘤分期、年龄、性别和淋巴结状态[15]),并对每个X进行标准化,即X服从均值为0和方差为1的标准正态分布,ti表示个体i的生存时间,样本量设为n。建立如下的一般Cox模型[24]:
(1)
其中h0(t)是所有协变量取值为0时的基线风险函数,a=(a1,a2,…,ap)是临床协变量的系数。
4.具有临床协变量和基因表达水平的稀疏Cox模型
设Gi为m维向量,表示个体i的一组遗传标志物(如TCGA数据中基因表达水平),并对每个标志物进行标准化,即G服从均值为0和方差为1的标准正态分布,包含Xi和Gi的Cox模型为:
(2)
其中b=(b1,b2,…,bm)为遗传标志物效应大小的m维向量。在高维的背景下,基因数量远远大于样本大小(即m≫n),所以传统偏似然估计已不适用。在过去的几年里,出现了许多惩罚来估计未知参数的正则化方法[25-27]。本文使用lasso降低数据维度[27],lasso利用系数的绝对值函数来完成对模型系数的压缩,将一些基因表达的系数收缩为零以同时执行特征选择[28]。具体来说,lasso符合以下Cox模型(用coxlasso表示):
(3)

5.具有临床协变量和基因表达水平的线性混合Cox模型
如前所述,正则化方法(如coxlasso)本质上是稀疏模型,它假设只有少数基因与生存相关。然而线性混合Cox模型(coxlmm)将所有基因纳入模型中[12,31-32],则可认为coxlmm是多基因模型,因为其明确假设所有基因都参与了疾病的进展,并且对生存均具有影响[33-34]。在coxlmm模型中假设基因效应服从正态分布[35]
(4)
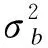
(5)
基于上述关系构造了一个新的等价的Cox混合模型:
(6)
其中Zi是K1/2和K=GGT的第i行向量,在遗传预测中通常K被称为遗传相关矩阵[12-13];δ是Zi效应大小的n维向量。在变换之后,随机效应的维数从m减少到n。因为在TCGA和GEO数据集中n通常比m小得多,这使得coxlmm的计算效率大大提高。最后b的估计值为:
(7)
6.TCGA中膀胱癌临床和转录组信息的相对重要性
为了揭示临床和转录组信息在膀胱癌生存变异中的不同作用,需要量化其对生存表型的重要性[39-41],我们首先对风险函数进行对数变换,然后定义两个变量PCE和PGE[35]
(8)
前者代表有多少生存变异可以被临床信息单独解释,后者揭示有多少生存变异可以用转录组的信息来解释,其中var(x)是x的方差,通过E(x-E(x)2)/n计算,E(x)是x的期望,var(e)表示除X和G之外所有未解释部分的方差。PCE和PGE的总和可以看作是模型中现有临床和转录组信息共同解释的生存方差(PVE)的比例。我们使用Jackknife方法来得到PCE或PGE的95%置信区间[42]。
7.模型评价
本研究使用一致性指数(C-index)来评估模型预测的准确性[43]。C-index为0.5表示完全不一致,说明模型预测能力不强;而C-index为1表示完全一致,表明模型的预测结果完全符合实际情况。参照先前的工作[13],本研究使用蒙特卡罗交叉验证(Monte Carlo cross validations,MCCV)评估预测性能:首先将训练组患者(训练集的80%)和试验组患者(训练集的20%)从训练集中随机抽取100次,然后测试组用于验证之前使用训练组估计的模型。在本文的训练集预测分析中,与coxlasso模型相比,coxlmm模型通过整合全基因表达数据显示出了更加稳健的预测性能。然而,coxlmm性能的提高是否与加入了有用的基因表达数据有关仍是未知的。因此,本研究在外部验证数据集上计算C-index来进一步评估coxlmm的预测性能。
结 果
1.训练集中Cox、coxlasso和coxlmm模型的预测能力
在TCGA训练集中,Cox、coxlasso和coxlmm的C-index分别为0.652、0.655和0.676(图1)。在三个合并数据训练集中,Cox、coxlasso和coxlmm的C-index分别为0.651、0.650和0.671(图2)。在GSE31684训练集中,Cox、coxlasso和coxlmm的C-index分别为0.553、0.550和0.553(图3)。在大多数情况下与另外两个模型相比,coxlmm预测性能最好。例如,当TCGA数据集为训练集时,coxlmm的预测精度比Cox平均提高2.4%;比coxlasso平均提高2.1%。当三个合并数据集为训练集时,coxlmm的预测精度比Cox平均提高2.0%,比coxlasso的预测精度平均提高2.1%。然而当GSE31684数据集为训练集时,coxlmm与Cox的预测精度相同,coxlmm的预测精度比coxlasso平均高出0.3%。
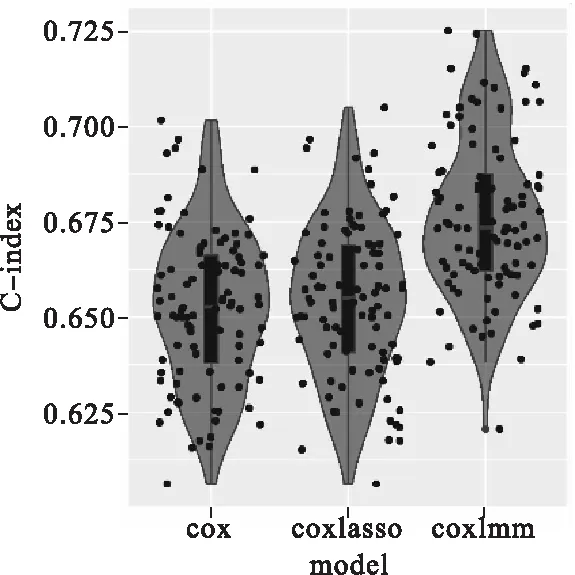
图1 TCGA训练集中的蒙特卡罗交叉验证(MCCV)
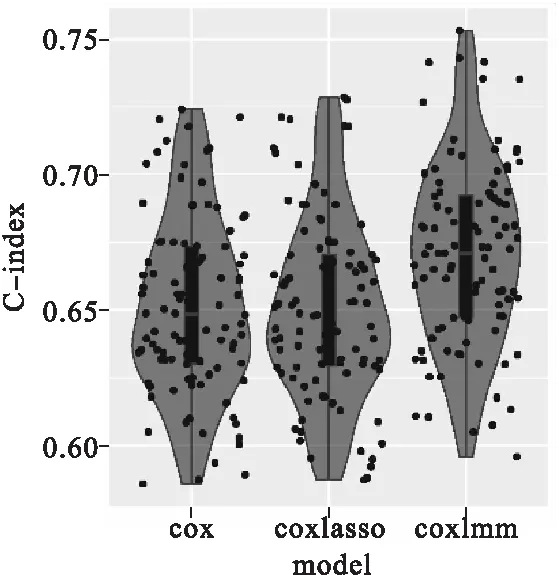
图2 三个合并数据训练集中的蒙特卡罗交叉验证(MCCV)
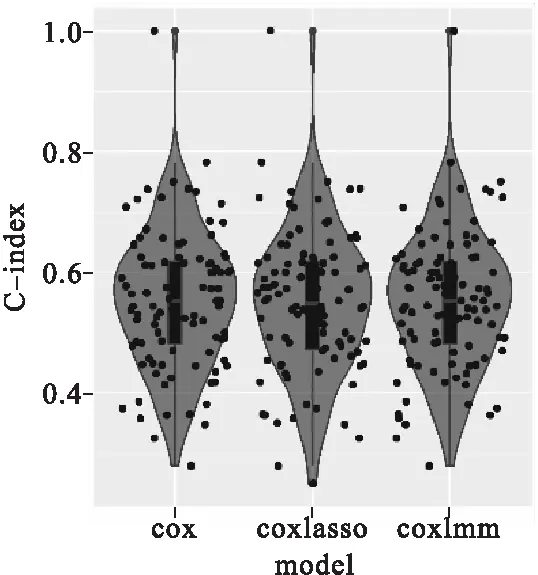
图3 GSE31684训练集中的蒙特卡罗交叉验证(MCCV)
2.验证集中Cox、coxlasso和coxlmm的预测能力
本文在使用外部数据集的情况下通过计算C-index来进一步评估coxlmm对预后的预测能力(表2),结果显示coxlmm比其他模型的预测精度更高。例如,当TCGA数据集作为训练集同时合并GEO数据集作为验证集时,coxlmm的C-index为0.534,coxlasso的C-index为0.527,coxlmm比coxlasso的预测精度提高0.7%,具有更强的预测能力;当GSE31684作为训练集,同时TCGA和GSE13507合并的数据集作为验证集时,Cox、coxlasso和coxlmm的C-index分别为0.624、0.632和0.633,coxlmm的预测精度比Cox和coxlasso分别提高0.9%和0.1%,同样具有更强的预测能力。三种模型在使用两种外部验证集的情况下,C-index结果如表2所示。

表2 在验证集中三种模型的一致性指数(C-index)
3.TCGA膀胱癌中估计PCE和PGE
本研究定义PCE和PGE以揭示临床和转录组信息在膀胱癌生存变异中的不同作用,TCGA中膀胱癌数据集PCE和PGE的估计值分别为14.95%和10.88%,其中PCE的95%置信区间为8.76%~24.35%,PGE的95%置信区间为3.98%~6.42%,PCE和PGE的总和PVE为25.8%。本研究中膀胱癌PCE的估计值略高于PGE的估计值。PVE较大(例如大于10.0%)表明临床信息和转录组信息在生存变异中均起重要作用;而本研究中膀胱癌的PVE为25.8%,结果表明临床信息和转录组信息在膀胱癌生存变异中均起重要作用。
讨 论
本文重点研究了三种方法(一般Cox模型、coxlasso模型和coxlmm模型)在TCGA和GEO膀胱癌数据集中对预后的预测效果,并系统地评估了转录组的预后价值[15-17]。研究结果显示,coxlmm模型比其他两种模型有更好的预测性能,表明全面整合临床和转录组信息可以提高预测的准确性。换言之,在预测模型中纳入所有基因比只纳入少部分基因具有更好的预测精度。本文利用TCGA数据集进一步评估了coxlmm模型中临床协变量和基因表达对膀胱癌生存变异的贡献。先前的研究表明,将除了基因表达之外的其他基因组测量数据结合到临床协变量中不能获得显著的功效增益[44],所以PCE和PGE能够涵盖几乎所有可用信息解释的生存变异。当转录组信息在PGE量化的变异中占很大比例时,使用基因表达信息的预测模型的准确性将得到显著提高。需要说明的是,本文的主要目的不是探讨如何有效地将多个组学数据整合到膀胱癌预测模型中[6,45-48]。相反,而是利用单个组学数据(即基因表达)通过比较coxlasso和coxlmm预测精度的差异来实现更好的预后预测。此外,将本研究的分析方法用于膀胱癌其他组学数据也是可行的。
本研究存在以下不足,首先,虽然coxlmm模型比其他两种模型的预测性能更好,但是C-index结果显示提升的精度并非特别明显。其次,本研究在分析时,只纳入了转录组信息而忽略了其他组学数据集(如拷贝数变化和DNA甲基化)可能会影响预测精度。由于在TCGA和GEO数据库中与膀胱癌有关的临床信息(如吸烟)缺失严重,不能将有效的临床信息纳入模型,这也可能会导致模型的预测性能被低估。再次,膀胱癌的有效样本量比较少,TCGA和GEO中的截尾数据的占比都比较高[15-17],所以不可避免地降低了模型预测的准确性,进而会影响预测模型在实践中应用的效果。另一个不可忽视的问题是,外部验证的样本量仍然较少,因此进一步使用更大的样本量外部验证模型是必要的。最后,TCGA和GSE31684数据集的种群都是欧洲人群,而GSE13507数据集的种群为亚洲人群,不同种群之间的差异可能也会影响模型预测的准确性。
本研究也有以下几个优势。首先,在coxlmm模型中整合全转录组数据同时联合临床信息对膀胱癌预后进行预测。其次,本研究中多个数据集的交叉验证以及外部验证保证了coxlmm模型预测性能的稳定性。第三,coxlmm是一种基于机器学习并且考虑了线性核的预测方法,它为进一步研究如何改善预后评价提供了可能。
我们注意到在其他现有的高维数据统计分析方法中,随机森林也用于生存结局的预后预测[49]。随机森林具有抗噪声、防止过拟合、可处理非线性相关等优点,在基因表达数据判别分类研究中有着较好的分类性能,但大量的噪声仍会影响随机森林的分类效能,例如在基因表达差异很大的情况下,随机森林中的每棵树的节点很少,极有可能使其他差异基因不被抽到,从而漏掉相当一部分有差异的基因[50]。也即是,当基因表达差异很大时,coxlmm模型的预测效能可能优于随机森林方法。然而,当生存数据删失比例较大时,随机森林和coxlmm模型可能均达不到预期的效果。因此,未来有必要进一步在高维数据背景下比较coxlmm模型与其他方法的优缺点。
最后,我们意识到混合模型(mixed models)和稀疏模型(sparse models)的预测效果优劣取决于对基因与疾病关系的假设与真实情况是否吻合。然而,实际中基因与疾病之间真实的关系往往未知。在今后的研究中,我们可以在遗传预测的基础上,结合这两个模型构建一种混合稀疏预测方法[13,34],混合稀疏Cox模型有望具有更好的预测性能。
综上所述,在预测膀胱癌预后时,与仅纳入少部分转录组学信息相比,纳入全转录组信息可以提高预测能力。这些也为更深入地了解膀胱癌肿瘤进展机制提供了机会。
