T幼淋细胞白血病小细胞变异型2 例临床分析
2022-03-16王菱菱陆米则
王菱菱,王 珏,陆米则,程 枫,杨 蕾,周 新,陆 焱,钱 瑛
(1.南京医科大学附属无锡人民医院血液科,江苏 无锡 214023;2.南京医科大学附属无锡人民医院医学检验科,江苏 无锡214023)
T幼淋巴细胞白血病(T-cell prolymphocytic leukemia, T-PLL)是一类罕见的临床具有高度异质性的成熟T细胞肿瘤,其病程呈侵袭性,治疗效果差,生存时间短[1]。T-PLL约占幼淋巴细胞白血病中的20%,表达成熟胸腺后T细胞表型,主要临床特点为幼淋巴细胞异常增多,淋巴结肿大,肝脾肿大,多浆膜腔积液等。T-PLL临床表现及实验室检查易与其他成熟T细胞肿瘤混淆,如:Sezary综合征、T细胞大颗粒淋巴细胞白血病、成人T淋巴细胞白血病等。临床医生通常5~10 年才诊治1 例T-PLL患者,极低的发病率使得临床医生很难全面认识这类疾病,且没有特异的形态学及其他实验室检查用于确诊T-PLL。因此综合分析临床表现、细胞形态学、免疫分型、分子生物学、细胞遗传学是诊断T-PLL的主要手段[2]。本文我们报道2 例T-PLL小细胞变异型患者的临床与实验室检查资料,并对相关文献进行复习。
1 临床资料
病例1,男性,54 岁,因“胸痛一周”于2020年12月09日入院。入院查体:体温37.5 ℃,脉率80 次/min,呼吸17 次/min,血压139/84 mmHg;神志清楚,轻度贫血貌,皮肤黏膜未见瘀斑瘀点,无黄染;颈部,腋窝,腹股沟处可触及多枚淋巴结肿大,直径1~2 cm,质韧、无触痛、无融合、活动可,胸骨无压痛,双肺呼吸音粗,未闻及干湿性啰音;心律齐,未闻及病理性杂音;腹平坦,无压痛及反跳痛,肝肋下未及,脾脐下1 cm,质硬,无压痛,移动性浊音阴性,双下肢无水肿。血常规示:WBC 43.83 ×109/L、淋巴细胞计数39.23×109/L、HGB 102 g/L、PLT 48 ×109/L。外周血形态:不典型淋巴细胞占81%,该类细胞胞体小,胞质少见,蓝色,无颗粒,核不规则,异形性明显,胞核多突起,核染色质聚集,部分细胞隐约可见核仁(图1A)。血生化检查:血糖6.94 mmol/L、直接胆红素7.3 μmol/L、天门冬氨酸氨基转移酶(AST)47 U/L、乳酸脱氢酶(LDH)331 U/L、腺苷脱氨酶29.5 U/L。β2微球蛋白4.12 mg/L。淋巴细胞亚群:总T淋巴细胞(CD3+):95.32%、辅助T细胞(CD3+CD4+):65.67%、细胞毒T细胞(CD3+CD8+)4.96%、B淋巴细胞(CD19+)3.81%、NK细胞(CD3-CD16+CD56+)0.43%。骨髓形态:增生活跃,粒红比1.27:1,成熟淋巴细胞异常增生,约占87%,该细胞胞体小,胞浆少见,嗜碱性,部分细胞胞核不规则,可见扭曲或突起,核染色质较粗糙,少数细胞隐约可见核仁(图1B)。骨髓活检病理:CD4+CD8-T淋巴细胞淋巴瘤侵犯骨髓。HE及PAS染色示送检骨髓增生极度活跃(约90%),小淋巴细胞弥漫浸润,各阶段粒细胞红细胞散在分布,巨核细胞少见。网状纤维染色(MF-1级)(图1C、1D)。骨髓流式细胞术检测:异常T淋巴细胞表型细胞群占有核细胞的52.96%,表达CD3、CD5、CD2、CD4、CD45RO,弱表达CD7、CD161,不表达TCRγ/δ、CD8、CD10、CD19、CD20、CD57、CD56、CD16、CD25、CD94、Perforin、GranzymeB、CD45RA、CD30(图2)。T细胞受体(TCR)γ基因重排阳性,TCRβ基因重排阳性,TCRδ基因重排阴性(图3)。染色体核型:46,XY[20]。右侧颈部淋巴结活检:CD4+CD8-T细胞淋巴瘤。免疫组化:CD3+,CD5+,CD2+,CD7+,CD20-,PAX5-,CD10-,CyclinD1-,Ki67阳性率约10%,BCL2-,CD56-,CD57-,TIA1-,CD4较多+,CD8部分+,CD138-,CD30-,CD25-,ALK-。荧光原位杂交(FISH):EBER-。二代基因测序(NGS):STAT5B 3.0%,JAK3 26.70%,ATM(p.G2765C)73.80%,ATM(p.E2768A)73.0%。颈胸腹部位CT:颈部散在稍大淋巴结,脾大,腹腔及腹膜后多发肿大淋巴结,两侧腹股沟多发稍大淋巴结。诊断符合T-PLL。患者依从性差,拒绝接受化疗,要求出院,失访。
病例2,男性,81 岁,因“颈部多发淋巴结肿大五月余,加重一周”于2019年01月25日入院。入院查体:体温36.5 ℃,脉率103 次/min,呼吸18次/min,血压140/78 mmHg;神志清楚,轻度贫血貌,皮肤黏膜未见瘀斑瘀点,无黄染;颈、腹股沟处可触及多枚淋巴结肿大,直径1~3 cm,质韧、无触痛、边界清晰、活动可,胸骨无压痛,双肺呼吸音清,未闻及干湿性啰音;心律齐,未闻及病理性杂音;腹平坦,肝肋下未及,脾肋下三指,质硬,无压痛,移动性浊音阴性,双下肢无水肿。血常规示:WBC 134.38 ×109/L、淋巴细胞计数55.23 ×109/L、HGB 116 g/L、PLT 14 ×109/L。血生化检查:血糖6.94 mmol/L、总胆红素37.2 μmol/L、AST 55 U/L、LDH 1 537 U/L、腺苷脱氨酶115U/L,尿素8.5 mmol/L,尿酸480.7 μmol/L。β2微球蛋白8.07 mg/L。淋巴细胞亚群:总T淋巴细胞(CD3+):95.9%、辅助T细胞(CD3+CD4+):94.9%、细胞毒T细胞(CD3+CD8+)0.9%、B淋巴细胞(CD19+)0.8%、NK细胞(CD3-CD16+CD56+)3.2%。外周血形态:淋巴细胞体积小,胞浆少见,蓝色,无颗粒,核型欠规则,部分细胞可见扭曲,核仁不清楚。骨髓形态:有核细胞增生明显活跃,全片淋巴细胞比例明显增高,占85%,成熟淋巴细胞占多数,胞体小,胞浆少见,蓝色,部分细胞核型不规则,可见扭曲或突起,核染色质较粗糙,核仁不清楚;粒系比例减低,占8.5%,红系占5%;巨核细胞全片1 只,血小板少见,意见:慢性淋巴增殖性疾病。骨髓活检:HE及PAS染色示骨髓增生极度活跃(80%),淋巴细胞增多,胞体大小不一,染色质细致,粒红系细胞散在分布,巨核细胞数量大致正常。网状纤维染色(MF-1级)。免疫分型:异常T淋巴细胞表型细胞群占有核细胞的88%,表达CD3、CD5、CD2、CD4、HLA-DR、CD45,弱表达CD7,不表达CD8、CD10、CD19、CD20、CD22、CD57、CD56、CD16、CD13、CD33、CD138、CD38。TCRγ基因重排阳性,TCRβ基因重排阳性,TCRδ基因重排阴性。染色体核型:46,XY[2]。FISH:TP53基因缺失阳性(42%),CEP12、D13S319、ATM基因未见异常,CCND1/IGH融合基因阴性。NGS:TP53基因编码序列发现p.V203L突变,突变频率44.6%。胸腹部CT:两侧胸膜增厚,两肺多发陈旧性病灶,纵膈及两侧腋下多发肿大的淋巴结;脾大,左侧肾上腺增粗,腹膜后多发肿大淋巴结。患者不接受化疗,出院后口服激素治疗,随访约1 个月后死亡。
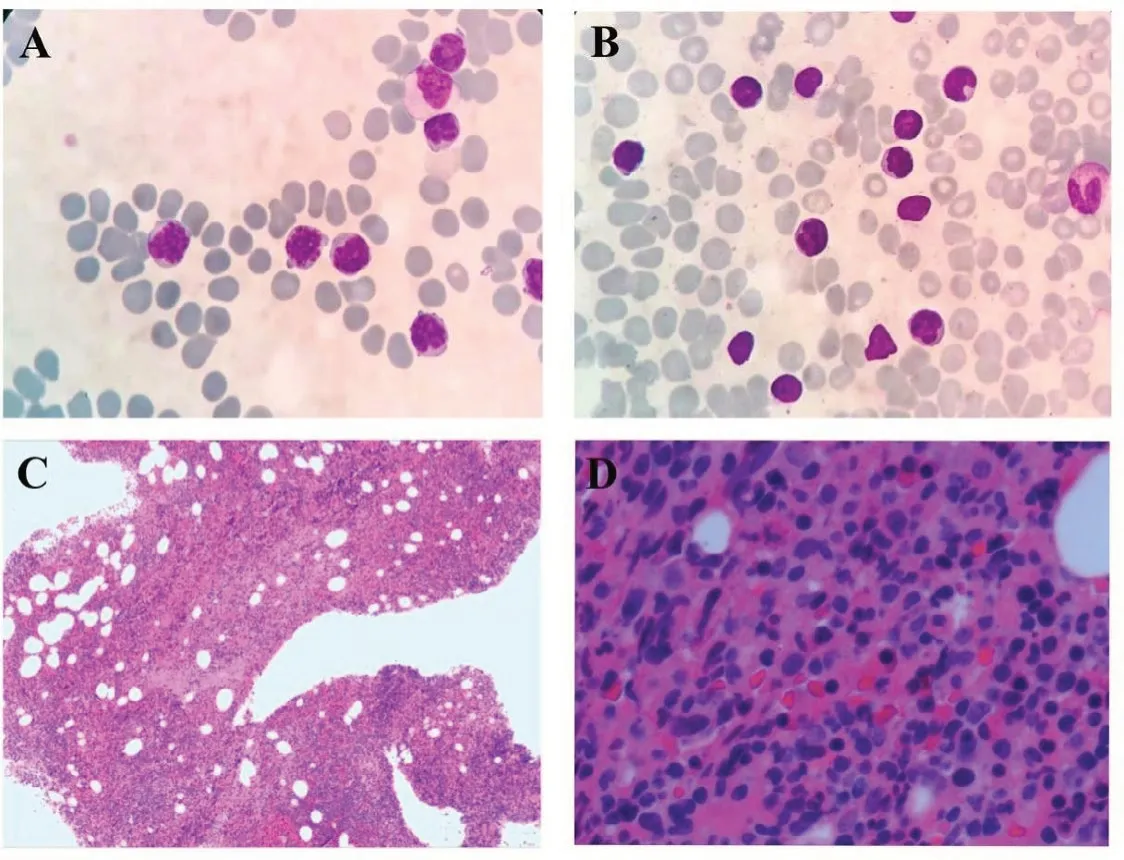
图1 形态学结果
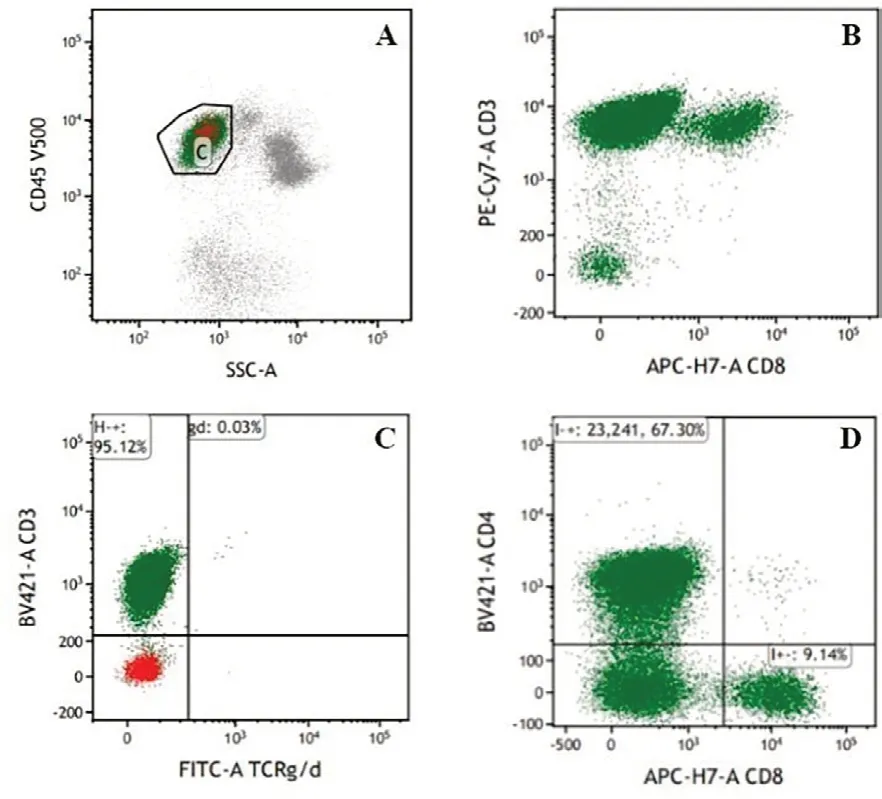
图2 流式免疫分型结果
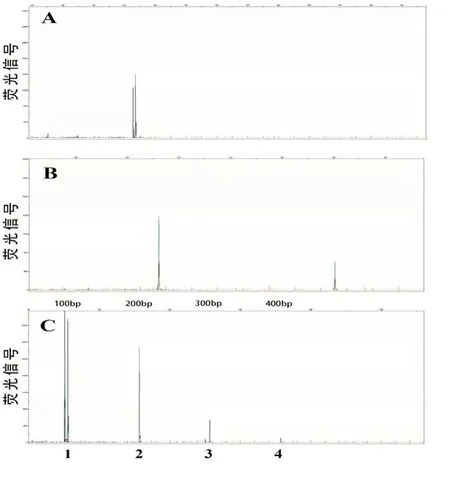
图3 毛细血管电泳法检测TCR基因重排结果
2 讨论
T-PLL是一种极罕见的T细胞性肿瘤,1973年首次报道该疾病,多见于老年人,中位发病年龄61岁,男女比例约为2:1。约15%患者起病时呈惰性经过,但维持一段时间后疾病终将进展,且一旦疾病进展,极为迅猛,预后差[3]。T-PLL相关的研究多为病例报告、单中心回顾性研究和小规模的临床研究,这些研究对于T-PLL诊断标准及疗效评估标准不一致,所以很难进行系统性分析总结[4-6]。T-PLL在临床表现和实验室检查结果与慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)不同,而早期T-PLL的诊断标准主要依据国际慢淋工作组(IWCLL)对CLL的诊断标准,但是CLL的诊断标准并不总是适用于T-LL[7]。2019年,T-PLL国际研究组就本病提出了新的诊断标准[主要诊断标准:淋巴细胞>5 ×109/L,具有典型的T幼淋巴细胞异常免疫表型;异常克隆T淋巴细胞,通过PCR或流式细胞术方法检测TCR基因重排证实;典型的遗传损害(TCL1A/B或MTCP1改变或14号染色体异常)。次要诊断标准:11号染色体异常(11q22.3;ATM);8号染色体异常:idic(8)(p11), t(8;8),trisomy 8q;5号,12号,13号,22号染色体异常或者复杂核型;临床有T-PLL特定部位受累(如脾肿大、积液)],确诊T-PLL需符合三个主要诊断标准,或满足前两个主要诊断标准和一个次要诊断标准,也可以诊断为T-PLL[8]。
T-PLL患者外周血白细胞计数显著增多,常>100 ×109/L,幼淋巴细胞≥55%,约半数病例出现贫血和血小板减少。生化检查常发现LDH及β2微球蛋白水平升高,无高钙血症。血清学或聚合酶链反应方法检测人类嗜T细胞病毒(HTLV)Ⅰ型和Ⅱ型始终为阴性,提示该逆转录病毒与T-PLL的发病机制无关[9]。本组2 例外周血淋巴细胞计数明显升高,且出现不同程度贫血及血小板减少,LDH及β2微球蛋白水平升高。
外周血细胞形态对T-PLL的诊断最为关键,可以表现为3种形态异常[10-11]。75%为经典型,特点是T-PLL细胞中等大小,核圆形,椭圆形或不规则形,核染色质中等致密,可见明显的中位核仁,胞质嗜碱性,无颗粒;20%为小细胞变异型,细胞体积较小,胞质少,细胞核不规则,核仁不明显;5%为脑回型,与Sezary细胞类似,细胞核似脑回,胞质泡样突起。本组2 例外周血形态特点为细胞体积较小,核异形性较明显,核仁不明显,符合小细胞变异型T-PLL形态特征。其他组织(如骨髓和淋巴结)的细胞形态学检查可能无法明确区分T-PLL和其他成熟T细胞肿瘤,因此不是确诊T-PLL必需的检查,但骨髓形态学检查对治疗后评估疾病缓解程度必不可少。本组例1骨髓活检和淋巴结活检均提示CD4+CD8-T细胞淋巴瘤,而例2骨髓形态学结果提示慢性淋巴增殖性疾病,并无法明确区分T-PLL和其他成熟T细胞肿瘤。
免疫分型检查对T-PLL和其他成熟T细胞肿瘤鉴别诊断意义较大[12-13](表1)。T-PLL起源于胸腺后T细胞,无T系前体细胞标记,如CD34、CD1a、TdT,无NK细胞标记,如CD16及CD56,表达泛T细胞标记,如CD2、CD3、CD5、CD7在T-PLL强表达,但也可见弱表达现象。CD4+CD8-最常见,也可见CD4+CD8+及CD4-CD8+[14]。本组2 例患者免疫表型均为CD4+CD8-,表达泛T细胞标记CD2、CD3、CD5、CD7,且不表达CD16、CD56、CD1a、TdT,符合T-PLL免疫表型特点。

表1 T-PLL的免疫表型及相关疾病的鉴别诊断
细胞遗传学提示T-PLL病例中T细胞受体基因(TCRβ或TCRγ)存在克隆性重排(经PCR/NGS或流式细胞术方法证实)。T-PLL病例常检测出复杂染色体核型(70%~80%),所以染色体显带分析和FISH可以为T-PLL与其他T淋巴细胞增殖性疾病提供依据[15]。T-PLL病例常有8、11、14、17、X染色体异常,导致MYC(位于8q24)、ATM(位于11q22.3)、TCL1(位于14q32)、TP53(位于17号染色体)、MTCP1(位于Xq28)基因的表达异常。在80%~90%的T-PLL病例中检测到11q23(ATM)位点的突变,相关研究[16]证实在ATM基因异常的背景下,TCL1过表达具有促白血病的协同作用。Bergmann等[17]研究发现在31%的T-PLL病例中检测到TP53基因缺失,而14%的病例存在TP53基因突变,T-PLL病例伴TP53基因缺失或突变均提示疾病预后差。Stengel等[18]应用二代基因测序技术检测T-PLL病例,发现约75%的病例存在JAK/STAT通路的激活,且T-PLL病例伴JAK3基因突变提示疾病预后差。本组2 例TCR基因重排均为阳性且染色体核型为正常核型。例1通过二代基因测序检测发现有STAT5B、JAK3、ATM基因突变;例2通过FISH检测发现TP53基因缺失阳性,二代基因测序结果提示TP53基因突变。本组2 例细胞遗传学检查结果与目前国外文献报道结果基本一致,T-PLL病例存在TP53基因异常表达,提示预后不佳,生存期短,本组例2 有TP53基因异常表达,生存期仅1 个月。
T-PLL属于罕见病例,目前尚无治疗指南。阿仑单抗是治疗T-PLL效果最佳的药物,它是一种针对表面CD52抗原的完全人源化的IgG1抗体,几乎所有的T-PLL细胞表面都表达CD52,Dearden等[4,19]报道应用阿仑单抗治疗T-PLL总体反应率(ORR)高于90%,无进展生存期(PFS)在8~11 个月。尽管ORR较高,但患者仍会复发,有应答者缓解的中位持续时间少于2 年。因此,完全缓解的患者应考虑进行异体干细胞移植(allo-HSCT)巩固治疗。尽管治疗相关死亡率和复发率仍然很高,但大约1/3的患者可以通过allo-HSCT获得长期生存[20]。欧洲血液和骨髓移植学会的一项前瞻性研究[21]结果表明,接受包含至少6 Gy全身照射作为移植预处理的患者复发率较低,对于无供体来源的患者,自体干细胞移植也是一种选择,可以明显延长PFS。复发后,T-PLL的预后很差,再次使用阿仑单抗仍有约50%的患者对其有反应,但总体PFS较短。通过体外药敏研究探索治疗T-PLL的新的靶向药物,新的药物如BCL-2抑制剂、HDAC抑制剂和JAK3抑制剂可能对T-PLL有治疗作用[22],将来需要更多的前瞻性临床研究来提高疗效。
