Analysis of key pathogenic target genes of ovarian cancer and experimental verification of cells in vitro
2022-03-16WANGYihanCHENBocenPENGYunhua
WANG Yi-han, CHEN Bo-cen, PENG Yun-hua✉
1.The Second Affiliated Hospital of Hainan Medical University, Haikou, 570216, China
2.Hainan Medical University, Haikou, 571199, China
Keywords:
ABSTRACT Objective: To mine genes highly related to the pathogenesis of ovarian cancer by using multichip integrated bioinformatics methods and verify them in cells, which provided key genes and important theoretical basis for targeted research of ovarian cancer.Methods: Three datasets,GSE38666, GSE40595 and GSE54388, were downloaded from the Gene Expression Integrated Database database for differential gene (DEGs) screening, including 26 normal samples and 65 ovarian cancer samples.Gene ontology functional annotation of selected DEGs was performed through DAVID online database to clarify the biological characteristics of DEGs.The main pathways of DEGs were obtained by enrichment analysis using Kyoto gene and genomic encyclopedia method.Based on the STRING database, the DEGs protein-protein interaction network was constructed by using CytoScape software, and the key genes were screened by GEPIA2 database to verify the expression at the cell level.Results: A total of 238 DEGs were screened from GSE38666, GSE40595 and GSE54388 datasets, of which 168 DEGs were upregulated and 70 DEGs were down-regulated.The co-expressed DEGs were mainly enriched in biological functions such as mitotic nuclear division, spindle, chromosomal region and DNA helicase activity in ovarian cancer.They were mainly involved in biological processes such as cell cycle, DNA replication, oxidative phosphorylation and biosynthesis of amino acids,thereby affecting the occurrence and development of ovarian cancer.Six genes were highly expressed and associated with the development of ovarian cancer, including IFI27, EPCAM,CXCR4, PEA15, CLDN3 and CAPG.Cell verification showed that the mRNA expression of the six genes in ovarian cancer cells was higher than that in normal ovarian cells (P<0.05),which was consistent with the previous screening results.Conclusion: Multi-chip integrated bioinformatics is an effective method to find ovarian cancer target genes.IFI27, EPCAM,CXCR4, PEA15, CLDN3 and CAPG are highly correlated with the occurrence and development of ovarian cancer, which can be used as target genes for ovarian cancer research.
1.Introduction
Ovarian cancer is a common female genital malignant tumor with the highest mortality rate, and the 5-year survival rate is still hovering at 49.7% (https://seer.canc-er.gov/statf- acts/html/ovary.html).The etiology and pathogenesis are unclear, early diagnosis is difficult, lack of sensitive and specific markers, chemotherapy multidrug resistance and recurrence rate is high, effective target molecule research is the focus of ovarian cancer research[1,2].Bioinformation technology can effectively integrate a variety of data sets and carry out in-depth mining, effectively screen out the key genes acting on the disease, and gradually become an important method for disease prevention, treatment and prognosis research[3-5].We screened through the comprehensive gene expression database (Gene Expression Omnibus, GEO), analyzed the gene by a variety of bioinformatics techniques, and verified it by cell experiments in vitro.In order to find the key target genes highly related to ovarian cancer and provide the basis for the targeted research of ovarian cancer.
2.Materials and methods
2.1 Experimental materials
Human ovarian cancer cell line SKOV3 was purchased from Shanghai Hongshun Biotechnology Co., Ltd.(Shanghai, China).Human ovarian epithelial (IOSE80) cells were provided by Shanghai Zeye Biotechnology Co., Ltd.(Shanghai, China).RNA extraction kit was purchased from Shanghai Plomag Biological products Co., Ltd.(Shanghai, China).Reverse transcription kit and real-time fluorescent quantitative PCR kit were purchased from Shanghai Yisheng Biotechnology Co., Ltd.(Shanghai, China).Fetal bovine serum was purchased from Yeasen Biotechnology Co., Ltd.(Shanghai, China).Trypsin was purchased from Gibco Company,USA.PBS was purchased from Hyclone Company, USA.McCoy's 5A medium was purchased from Gibco Company, USA.DMEM medium was purchased from BI Company, USA.
2.2 Acquisition of microarray data
The data sets GSE38666, GSE40595 and GSE54388 related to ovarian cancer microarray were found by GEO database (https://www.ncbi.nlm.nih.gov/geo/) for analysis.The genetic data of 26 normal ovarian samples and 65 ovarian cancer samples were included.GSE38666 includes 8 normal stroma and 8 matched normal ovarian surface epithelium in 12 individuals, 7 cancer stroma and 7 matched cancer epithelium in 18 patients with ovarian cancer.GSE40595 contains 31 samples of cancer-related matrix microdissected by laser, 32 samples of epithelial tumors from patients with high-grade serous ovarian cancer, 8 samples of microdissected normal ovarian matrix and 6 samples of ovarian surface epidermis (hoses).31 samples of laser microdissected cancer-related matrix and 8 samples of microdissected normal ovarian matrix were selected for the study.GSE54388 contains 16 high-grade serous ovarian cancer patients with laser microdissection epithelial tumor samples and 6 microdissection ovarian surface epithelium (hose) samples.The above data sets are based on GPL570, [HGU133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 arrays, series matrix files and platforms are downloaded as CEL files.
2.3 Identification and integration of DEGs
The differentially expressed genes between ovarian cancer and normal ovarian tissues from three microarray datasets were analyzed by R language limma package.The data of three chips are normalized by RMA algorithm.With P<0.05, |logFC|>1 for the filter screen for subsequent analysis of DEGs.The heat map is generated by HOTMAP 2 in GPLOTS package (https://cran.rproject.org/Web/Packages/gplots/).Wayne diagram was used to analyze the changes of differentially expressed genes (differentially expressed genes,DEGs) in the integrated data set.
2.4 GO and KEGG pathway enrichment analysis of DEGs
Visualization, annotation and integration discovery database(DAVID) contains high-throughput gene function analysis (https://david.ncifcrf.gov/).Proteins encoded by each candidate gene were enriched in pathway and function, and then annotated using DAVID database.Gene Ontology (GO) analysis is mainly used to effectively identify the biological characteristics of DEGs.Use the David Online tool to annotate DEGs.GO analysis includes three different categories : biological process (BP), cell component(CC) and molecular function (MF).For each GO term, the formula Fold Enrichment= (kink N) / (M hand N) is used to calculate the folding enrichment.The concentration analysis process of Kyoto Encyclopedia of Genes and Genomes (KEGG) method was completed, and the functional content of DEGs was obtained.Use Kobas online analysis database to complete the KEGG path analysis processing of DEGs (online available: http://kobas.cbi.pku.edu.cn).In this process, we compared and analyzed the significant upregulation and down-regulation of DEGs in detail based on the integrated microarray ovarian cancer data.
2.5 Network integration of protein-protein interactions
Use search tools to effectively search PPI networks constructed from gene databases (STRING, version 10.5, URL: https://stringdb.org/), and Cytoscape (v 3.8.2).Each node is a molecule,protein or gene, and each connection between nodes acts as a molecule to identify the internal pathways and functions of DEGsencoded proteins in ovarian cancer.The central node protein may be an important candidate gene or core protein with important physiological regulation function.
2.6 Cell culture
Before the experiment, the morphology and density were observed under microscope, and the medium in the original bottle was sucked out.Then 2 mL PBS was added for cleaning, and 1 mL 0.25%trypsin was added for static digestion for 1-2 min.Complete medium with the same volume as 0.25% trypsin was added for termination of digestion.The cells were transferred to the centrifuge tube and 5 min by 1 000 rpm centrifugation.After discarding the supernatant, the suspended cells in 3-4 mL complete medium were transferred to the culture flask and cultured in 5% CO237 ℃ cell incubator.
2.7 RNA extraction and real-time fluorescent quantitative PCR
Total RNA was extracted by RNA extraction kit and mRNA was reverse transcribed by reverse transcription kit.All operations are carried out according to the instructions for the use of the kit.The q225 fluorescence quantitative PCR instrument was used to detect the gene expression, and the reaction conditions were carried out according to the operation instructions of the fluorescence quantitative PCR kit (SYBR GreenMix, Roche).The thermal cycle parameters are as follows: 95 ℃ 300 s, then 95 ℃ 10 s, 60 ℃ 30 s,a total of 40 cycles.Three replicates were set for each reaction of quantitative PCR.GAPDH was used for mRNA internal reference.Data analysis using2-ΔΔCtmethod, Ct=experimental group (Ct target gene-Ct internal reference)-control group (Ct target gene-Ct internal reference).The amplified sequences of each gene and its internal reference are detailed in Table 1.
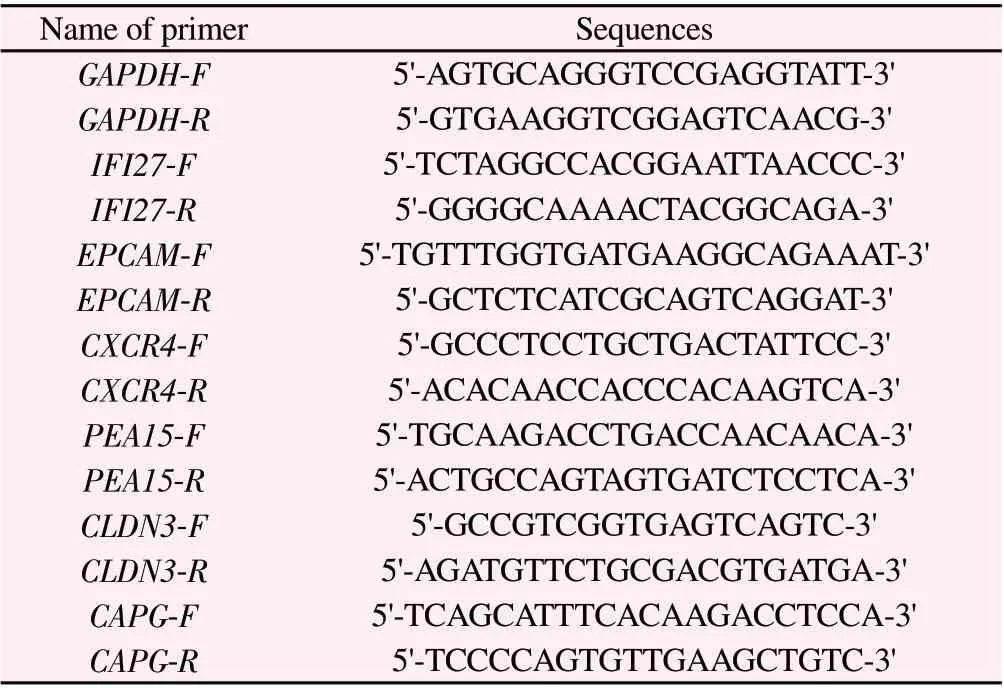
Tab 1 Primer sequences used in qRT-PCR
2.8 Statistical processing
GraphPad Prism 9.0 software was used to analyze all data.All data were repeated at least three times independently.Quantitative data were expressed as (±s).Independent sample t test or oneway analysis of variance were used to compare the homogeneity of variance between groups, otherwise nonparametric rank sum test was used.P<0.05 indicated that the difference was statistically significant.
3.Results
3.1 Microarray data information and analysis of DEGs in ovarian cancer
The mRNA microarray data related to ovarian cancer were searched in GEO database, and three DNA data GSE38666, GSE40595 and GSE54388 were selected for effective identification of DEGs.The three datasets were all generated by the same platform (GPL570,Affymetrix Human Genome U133 Plus 2.0 array).The gene normalization map of the dataset was obtained after standardization and other preprocessing, as shown in Figure 1.LIMMA software package screening (|logFC|>1, P<0.05).7 082 DEGs were selected from GSE38666 dataset, of which 5 205 were up-regulated and 1 877 were down-regulated; the GSE40595 dataset selected 4 239 DEGs, of which 3 095 were up-regulated and 1 144 were downregulated; the GSE54388 dataset selected 1 970 DEGs, of which 1 084 were up-regulated and 886 were down-regulated.Then, the top 200 clustering heats of gene analysis with the highest expression in each dataset are screened, and the clustering heat map of each dataset is obtained, as shown in Figure 2.David online analysis tool was used to complete data processing and analysis.238 DEGs genes that were uniformly expressed in three data sets were identified by Wayne diagram analysis, of which 168 were up-regulated and 70 were down-regulated, as shown in Fig 3.
3.2 Differential gene enrichment analysis of ovarian cancer
David online analysis tool was used to complete data processing and analysis, and P<0.05 was used as the criterion for enrichment analysis.GO functional annotation showed that the biological processes of 238 DEGs included mitotic sister chromatid separation,mitotic nuclear division and sister chromatid separation; cell composition was mainly enriched in spindle, chromosomal region and condensed nuclear chromosome kinetochore; Molecular functions include DNA helicase activity, single-stranded DNAdependent ATP-dependent DNA helicase activity and 3'-5' DNA helicase activity.Then through KEGG pathway enrichment processing online analytical database (http://www.Kegg.jp/kegg/kegg2.ht-ml), as shown in figure 4.KEGG pathway enrichment analysis 238 DEGs were mainly enriched in cell cycle, DNA replication, Oxidative phosphorylation, Biosynthesis of amino acids,ECM- receptor interaction, Thermogenesis, Relaxin signal pathway and so on, as shown in Figure 5.
3.3 Analysis of differential gene protein interaction network
The network diagram of 238 common genes was analyzed by STRING database and Cytoscape software.The PPI network expressed by DEGs in ovarian cancer was generated, and the nodes in the network represented proteins.The connection represents the interaction between proteins, as shown in Fig.6.Then it is verified by database GEPIA2 and literature search.Six key genes (IFI27,EPCAM, CXCR4, PEA15, CLDN3 and CAPG) were screened from 238 DEGs.

Fig 1 Gene expression normalization of ovarian cancer dataset

Fig 2 Hierarchical clustering heat map of ovarian cancer dataset DEGs

Fig 3 Wayne diagram of ovarian cancer dataset
3.4 The results of cell experiments in vitro
QRT-PCR verification of 6 key genes at cellular level.The results showed that the mRNA levels of IFI27, EPCAM, CXCR4, PEA15,CLDN3 and CAPG in SKOV3 cells were higher than those in IOSE80 cells.The difference was statistically significant (P<0.05), and the results were consistent with the previous analysis, as shown in Figure 7.

Fig 4 GO functional annotation of common differential genes in ovarian cancer dataset
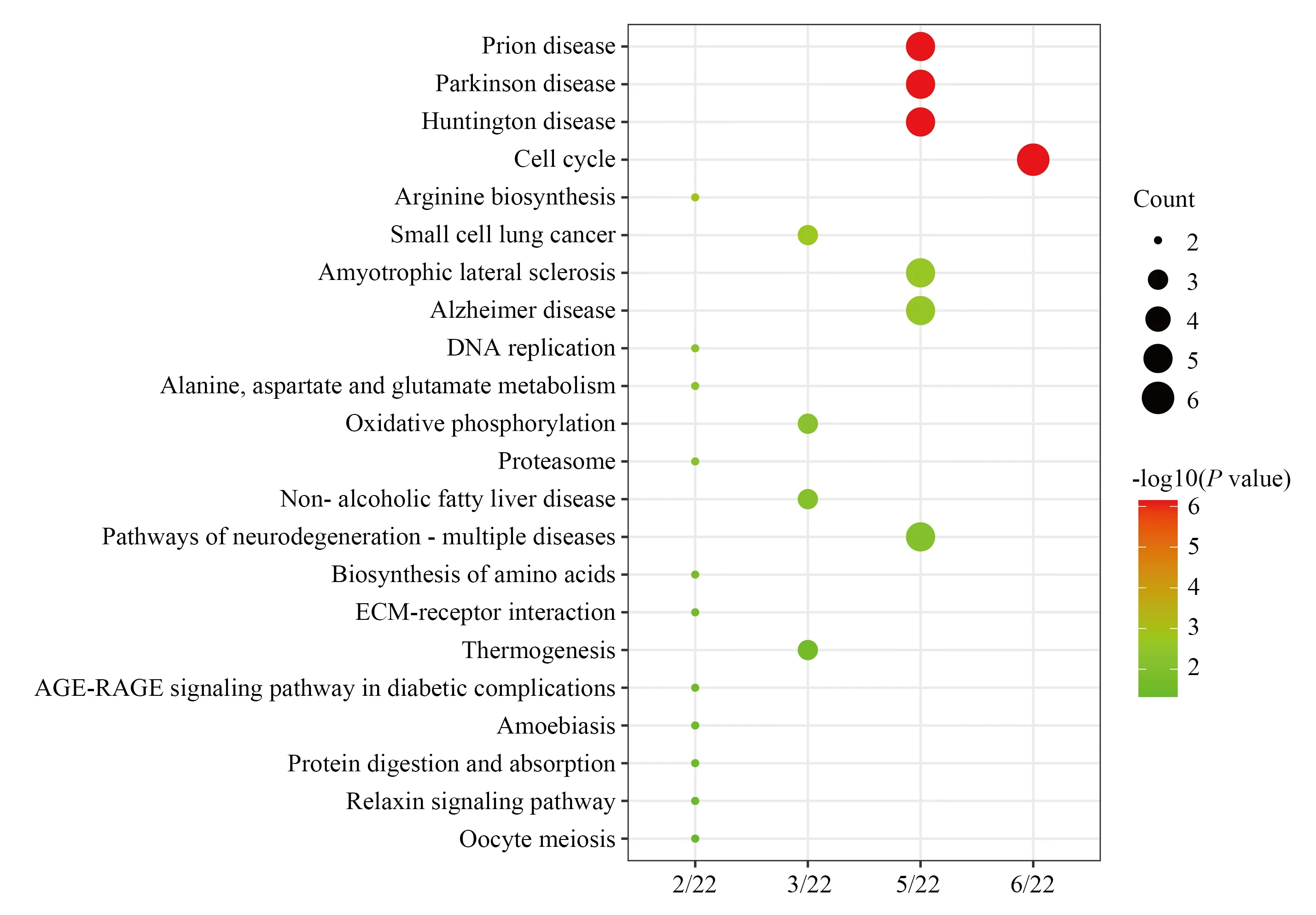
Fig 5 KEGG pathway enrichment analysis of common differential genes in ovarian cancer dataset

Fig 6 Protein interaction network analysis of differential genes
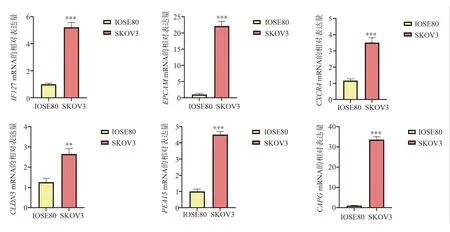
Fig 7 mRNA relative expression of key genes
4.Discussion
The application of bioinformatics technology has gradually become an effective pre-research method for tumor targeting research,which can provide important data information.In this study, three data sets GSE38666, GSE40595 and GSE54388 were integrated.Among them, 26 normal samples and 65 ovarian cancer samples were included.238 DEGs were co-expressed, of which 168 were up-regulated and 70 were down-regulated.GO enrichment analysis showed that it was mainly enriched in spindle, chromosomal region and condensed nuclear chromosome kinetochore, with DNA helicase activity, single-stranded DNA-dependent ATP-dependent DNA helicase activity and 3'-5' DNA helicase activity.It was involved in mitotic sister chromatid separation, mitotic nuclear division and sister chromatid separation.In this study, KEGG pathway analysis showed that these DEGs were mainly enriched in cell cycle, DNA replication, Oxidative phosphorylation, Biosynthesis of amino acids,ECM- receptor interaction, Thermogenesis, Relaxin signal pathway and other signal pathways.Among them, IFI27, EPCAM, CXCR4,PEA15, CLDN3 and CAPG were up-regulated in ovarian cancer.Further verified by cellular qRT-PCR experiment, the results showed that the expression of 6 key genes in ovarian cancer cells was higher than that in normal ovarian cells (P<0.05).Consistent with previous data and trends, it shows that it plays an important role in the development of ovarian cancer.
According to the literature review, interferon -inducible protein 27 (IFI 27) is located on human chromosome 14q32.Some studies have shown that IFI27 is highly expressed in ovarian cancer through bioinformatics, clinical case analysis and experimental verification from GSE58470, GSE45553, GSE41499, GSE33482 and GSE15372 data sets, and is related to platinum resistance in ovarian cancer[6].Li et al also showed that overexpression of IFI27 can promote epithelial-mesenchymal transition, cell migration and invasion and tumor growth in epithelial ovarian cancer[7].Epithelial cell adhesion molecule (EpCAM) is encoded by TACSTD1 gene on human chromosome 2.It is expressed on the basement membrane of most normal epithelial cells, not in non-epithelial tissues and squamous epithelial cells and some special epithelial cell types(such as epidermal keratinocytes, hepatocytes, thymic cortical epithelial cells) [8].It has been found that EpCAM can promote the proliferation, migration and invasion of ovarian cancer cells and has been used in clinical trials of various tumor immunotherapy, which is of great significance for clinical treatment of ovarian cancer[9,10].C-X-C motif chemokine receptor 4 (CXCR4) is a G protein-coupled receptor with seven transmembrane structures, and its coding gene is located on chromosome 2q21[11-13].CXCR4 specifically binds to its ligand CXCL12, which can activate a series of downstream intracellular signal transduction pathways and effectors, thereby regulating cell survival, proliferation, migration and adhesion[11-13].Zhang et al showed by bioinformatics that the higher the expression of CXCR4 in ovarian cancer, the shorter the progression-free survival and the shorter survival after progression, which is an important risk factor for advanced ovarian cancer[14].And cisplatin can increase the expression of CXCR4, promote the proliferation of tumor stem cells,enhance drug resistance, and form a vicious circle[15].A number of studies have shown that CXCR4-related drugs have great potential in clinical treatment, providing a new approach for tumors[16,17].Astrocyte phosphoprotein 15 (proliferation and apoptosis adaptor protein 15, PEA15) is a multifunctional small molecule phosphoprotein whose coding sequence is located on chromosome 1q21-22.Studies have found that the expression of PEA15 in ovarian cancer tissues is significantly higher than that in normal tissues by bioinformatics and experiments, which promotes the proliferation of ovarian cancer cells, suggesting poor prognosis, and is positively correlated with FIGO stage and histological subtype of ovarian cancer[18,19].Claudin 3 (CLDN3) is a member of the Claudin family and its coding sequence is located on chromosome 7q11.23[20].CLDN3 is highly expressed in various types of cancer[20].Huang et al have shown that CLDN3 can promote the migration, invasion and survival of ovarian cancer cells, and inhibit tumor growth after silencing its expression[21].Capping Actin Protein, Gelsolin-Like(GAPG) a member of the solgel protein superfamily, is an actinbinding protein encoded on chromosome 1q23.1.Some studies have found that CAPG is highly expressed in ovarian cancer, which can promote cancer cell migration and invasion, and can be used as a therapeutic target for ovarian cancer[22].Jiang et al through bioinformatics analysis, it was found that the expression of CAPG was positively correlated with regulatory T cell, tumor-associated and failure T cell infiltration in ovarian cancer, and negatively correlated with natural killer T cell and neutrophil infiltration[23].It can be used as a potential biomarker for judging the prognosis of ovarian cancer and the effectiveness of immunotherapy[23].
Therefore, the use of bioinformatics methods to integrate the data set of single or multiple diseases can effectively help in-depth study of diseases.In this study, six key genes were screened out through bioinformatics mining of ovarian cancer multi-chip data.The key target genes highly related to the pathogenesis of ovarian cancer were found by in vitro cell verification and related literature.It provides ideas and important theoretical basis for further in-depth study of ovarian cancer.
Author's contribution:
Paper design: Peng Yunhua, Wang Yihan; Bioinformation technology analysis : Wang Yihan, Chen Bocen ; Experimental operation : Wang Yihan ; Manuscript writing : Wang Yihan ; Article Review : Peng Yunhua.
All authors declare no conflicts of interest.
杂志排行

Journal of Hainan Medical College的其它文章
- Advances in pharmacological action of bergamot lactone
- Progress of improvement of pain and joint function of knee osteoarthritis treated with thunder-fire moxibustion in the last five years
- Abnormal expression of TGFβ1 in acute myeloid leukemia and its regulation effect on leukemia cells
- Investigation of paeonol-geniposide on acute alcoholic liver injury based on uniform design method
- Effect of Drynaria total flavonoids on the expression of NMDAR1,GluR2 and CaMK Ⅱ in the brain of hydrocortisone model mice
- Screening and functional analysis of the long-range interaction elements of β-globin genes