CCDC151基因新无义变异导致Kartagener综合征伴无精症表型
2022-03-14奚燕萍雷彩霞
肖 敏,奚燕萍,雷彩霞
(复旦大学附属妇产科医院上海集爱遗传与不育诊疗中心,中国 上海 200011)
纤毛的运动在身体中起着许多重要的作用[1]。以纤毛带动的黏液运动是呼吸道多纤毛上皮细胞表面协调运动的基础,形成黏液纤毛清除的主要宿主防御机制。在中枢神经系统中,脑脊液的流动是由位于脑室的纤毛调节的。在生殖系统中,输卵管纤毛帮助卵子向子宫推进;精子鞭毛在结构上与纤毛高度相关,驱动雄性配子运动。在胚胎发生早期,纤毛在左右不对称发育中发挥关键作用,它对于在侧板中胚层诱导不对称基因表达级联是必要的,决定了人体内脏器官在位置及形态上呈左右不对称分布[2]。
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD,MIM:244400)是一种遗传异质性常染色体隐性遗传为主的罕见纤毛病[3],发病率约1/20 000[4~7]。其特点是纤毛运动结构和功能异常,这种纤毛功能障碍可导致黏液纤毛清除缺陷,从而导致慢性上、下气道感染(如支气管炎和鼻窦炎),且近一半的病例会出现内脏转位。男性不育和女性生育能力降低在PCD中也很常见。PCD由于缺乏表征特异性,很难通过临床表型直接诊断。结合透射电子显微镜、高速视频显微镜和免疫荧光诊断工具分析患者纤毛形态和功能,是目前PCD诊断的主趋势。同时,基因检测也逐步被应用,因为迄今为止约有40个基因被鉴定与PCD有关[8]。中国人群关于PCD致病基因的研究报道开始于2007年,现在总约12篇涉及17例中国PCD患者基因突变的相关报道[9~11]。Kartagener综合征是PCD最常见的一种亚型,占PCD的40%~55%[4,6],因伴有内脏转位而归类命名。
CCDC151(coiled-coil domain containing 151),别名 MGC20983、ODA10(outer dynein arms 5),其编码的纤毛蛋白参与外动力蛋白臂组装和所需的运动纤毛功能,该基因是PCD致病基因之一,但国际范围内其突变的报道较少,中国人群仅发现1 例 CCDC151 c.167delG(p.G56Dfs*26)纯合突变[12]。本研究检测到中国人群纯合子CCDC151 c.1059C>A(p.Tyr353*)致病性变异1例,且首次报道了CCDC151基因变异导致无精子症不育表型;同时,总结了CCDC151基因突变谱,以利于后续研究。
1 材料与方法
1.1 患者资料
患者,男,29岁,夫妻双方染色体核型均正常,自述性生活规律,未避孕,未孕。患者家中独子,内脏反位,无精子症,AZF(azoospermia factor,Y chromosome gene)无缺失。外院诊断为梗阻性无精子症,考虑Kartagener综合征,患者父母为姨表亲。行睾丸切开取精术+输精管探查术+附睾取精术,精子冷冻保存备用于辅助生殖。本研究通过上海集爱遗传与不育诊疗中心伦理委员会批准,患者签署了相关知情同意书。
1.2 方法
1.2.1 DNA抽提和外显子捕获测序
按照凯杰DNA提取试剂盒标准操作流程从患者外周静脉血中分离基因组DNA(gDNA),采用Covaris技术将质控合格的gDNA样本随机打断成150~250 bp片段,并对DNA片段进行末端修复、加尾、连接接头及纯化。采用聚合酶链反应(polymerase chain reaction,PCR)对DNA片段进行扩增,并对扩增产物进行纯化。使用安捷伦Clear-Seq遗传性疾病试剂盒和SBS试剂盒进行外显子组杂交捕获。在Illumina HiSeq平台上进行全外显子测序。与人类参考基因组序列比对,95%的目标捕获区域的测序深度大于20X。医学相关外显子及上下游20 bp序列的平均测序深度≥94X,其中97%的目标序列的测序深度达20X以上。对所有测序片段进行碱基识别。二级分析流程主要采用Sentieon软件套装进行测序数据分析。测序片段通过Sentieon BWA与UCSC hg19参考基因组进行比对。
1.2.2 变异注释和筛选流程
变异采用VEP(Variant Effect Predictor)软件进行注释。每个碱基的测序深度及变异预测均从所有基因组测序数据中获得。三个主要的收录已知或疑似致病变异的数据库,包括ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)、人类孟德尔遗传在线(Online Mendelian Inheritance in Man,OMIM,https://omim.org/)和人类遗传疾病突变数据库(Human Gene Mutation Database,HGMD,http://www.hgmd.cf.ac.uk/ac/index.php),用于筛选已知的致病变异,同时采用多种工具预测错义变异的功能以及非编码调控序列的注释等。基于人群的大规模测序数据库用于排除在正常人群中具有较高频率的变异。需注意的是,变异注释过程使用的各公共或私有数据库定期进行更新,因此分析本例数据时,存在最新的文献证据未同步到相关数据库中的可能。拷贝数变异分析是基于高通量测序数据进行拷贝数变异分析测序,采用NextGENe软件(version 2.4.2.3)。变异致病性分类主要参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)和美国分子病理学会(Association for Molecular Pathology,AMP)、ClinGen(Clinical Genome Resource)变异解读工作组等发布的相关序列变异分类指南进行解读。基因命名采用国际人类基因组组织基因命名委员会(HUGO Gene Nomenclature Committee,HGNC)的命名规范。变异命名采用人类基因组变异学会(Human Genome Variation Society,HGVS)的命名规范。人类基因组参考序列版本为GRCh37/hg19。
1.2.3 Sanger验证
提取患者的外周血样本DNA。使用自动测序仪(3500XL Genetic Analyzer,Applied Biosystems Inc.,Foster City,CA,U.S.A.)从正向和反向两个方向对患者的变异进行Sanger测序验证。按照参考序列NM_145045.5(13个外显子)进行外显子编号。采用Primer Premier 5.0软件设计PCR扩增引物。Sanger测序的序列分析采用FinchTV软件(version 1.4.0)。Sanger测序所用引物为CCDC151-chr19-11534603-F1:AAACTGAACGGTAGCTGGAGGG和CCDC151-chr19-11534603-R1:CTTCCCTCCAGTTTGAGCCCTG。
2 结果与分析
2.1 先证者全外显子组测序及验证
基于患者的临床表型,选用以下人类表型标准术语(Human Phenotype Ontology,HPO)以筛选合适候选致病基因:HP:0012262:abnormal ciliary motility、HP:0001696:situs inversus totalis和 HP:0000027:azoospermia。经筛选,全外显子组测序结果显示,患者CCDC151(NM_145045.5)基因第8外显子 c.1059C>A(p.Tyr353*)纯合突变(图1),该突变可导致PCD 30型(ciliary dyskinesia,primary,30)[MIM:616037]:一种以运动纤毛异常为特征的疾病。由于呼吸道纤毛的缺陷,呼吸道感染导致慢性炎症和支气管扩张经常发生。由于胚胎淋巴结的纤毛功能障碍,患者可能表现出左右身体不对称和位置反转的随机化。经Sanger反向引物测序验证,该纯合变异真实存在(正向引物检测失败)(图2)。

图1 全外显子组测序点突变的可视化展示(Integrative Genomics Viewer,IGV)第一行是染色体位置坐标,标黄列表示突变位置。Fig.1 Integrative Genomics Viewer(IGV)for whole exome sequencing mutationsThe first row is the chromosome position coordinates,and the yellow column represents the mutation position.
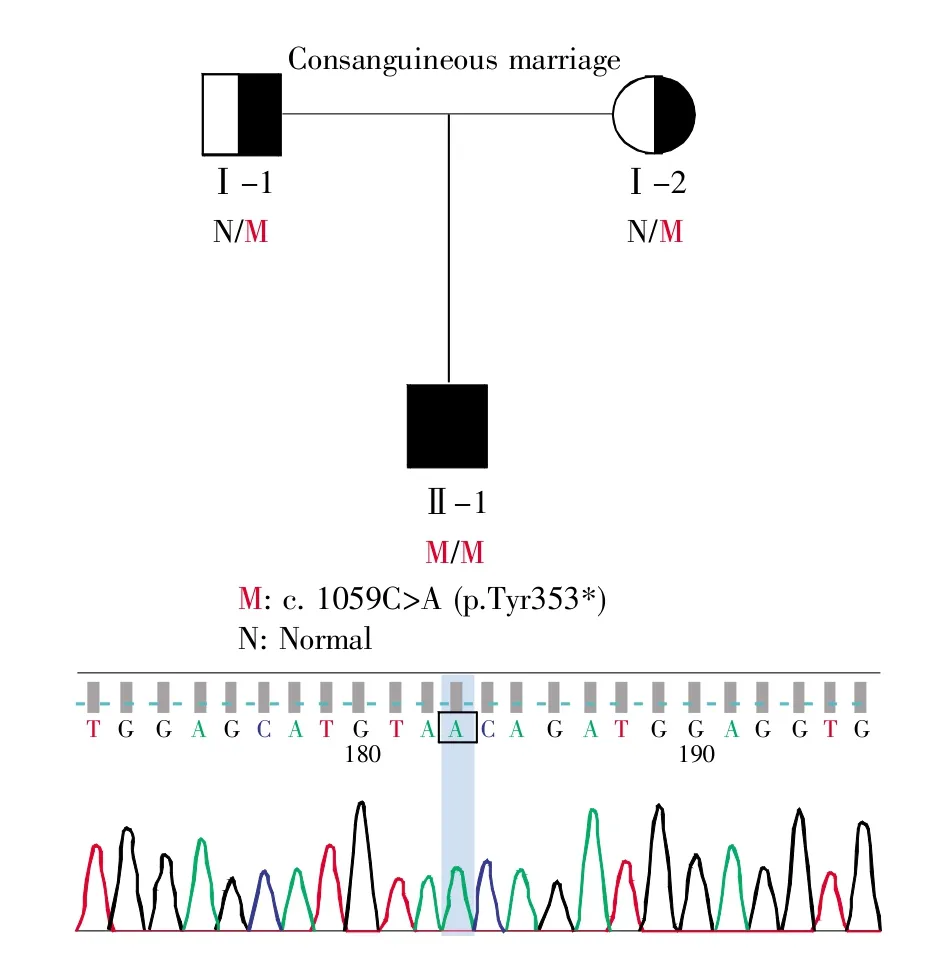
图2 患者CCDC151基因突变家系图及Sanger测序验证结果M表示携带c.1059C>A突变,N表示不携带c.1059C>A突变,蓝色柱状标记Sanger测序结果中变异所在位置。Fig.2 The family map of CCDC151 gene mutation of the patient and the Sanger sequencing verificationThe results show that M is carrying c.1059C>A mutation and N is not.The blue bar marks the location of the mutation in the Sanger sequencing results.
2.2 变异位点致病性解读
患者CCDC151基因在第8号外显子1 059位置的碱基从C转变到A,三联体密码由原来编码酪氨酸的密码子变成一个终止密码子,阅读框被一个过早的终止密码子打断,产生的mRNA将作为无义介导的mRNA被降解(nonsense-mediated mRNA decay,NMD)(图3)。根据ACMG指南,该致病性分类为致病(pathogenic,P)变异,证据如下:PVS1_Very Strong:在CCDC151基因中,功能丧失(loss-of-function,LOF)是一种已知的疾病机制(该基因有12个致病性的功能丧失变体),并且与PCD 30型(ciliary dyskinesia,primary,30)[MIM:616037]相关,完全符合当一个疾病的致病机制为功能丧失时,检出变异为无功能变异(无义突变、移码突变、经典±1或2的剪接突变、起始密码子变异、单个或多个外显子缺失);PM2_Supporting:隐性基因CCDC151在gnomAD外显子组数据库中未发现该变异(gnomAD外显子组覆盖率=76.3),在gnomAD基因组数据库中也未发现该变异(gnomAD基因组覆盖率=29.9),符合正常对照人群中未发现的变异(或隐性遗传病中极低频位点);PP3_Supporting:基于 BayesDel_addAF、DANN、EIGEN、FATHMM-MKL和MutationTaster等 5种致病性预测软件的分析均显示其是致病性的,无良性预测结果,符合多种统计方法预测出该变异会对基因或基因产物造成有害的影响;PP4:患者基因型与表型高度吻合,变异携带者的表型或家族史高度符合某种单基因遗传疾病。

图3 使用Mutalyzer软件(Mutalyzer 2.0.34,https://mutalyzer.nl)预测突变前后蛋白质编码的变化情况上图表示参考蛋白质序列的编码情况,红色氨基酸序列标记突变位置的参考氨基酸序列,下图是预测突变后的氨基酸编码情况,红色*号表示终止密码子。Fig.3 Changes in protein coding before and after mutation predicted by Mutalyzer software(Mutalyzer 2.0.34,https://mutalyzer.nl)The figure above shows the coding situation of the reference protein sequence.The red amino acid sequence marks the reference amino acid sequence from the mutation position.The figure below shows the coding situation of the predicted amino acid after mutation,and the red*represents the stop codon.
基于以上分析,我们认为c.1059C>A(p.Tyr-353*)纯合突变是患者Kartagener综合征伴无精子症的遗传学病因。
3 讨论
CCDC151基因位于19号染色体p13.2区域,编码一种螺旋-螺旋轴突蛋白质,可确保外动力蛋白臂复合体正确连接到微管卷曲-卷曲结构域[13]。在需要纤毛活动的物种中,该蛋白质高度保守。CCDC151是衣藻ODA10基因的脊椎动物同源体,ODA10基因在ODA相关的保守基因组装中发挥作用[14~15]。果蝇、斑马鱼和小鼠的功能分析表明,CCDC151参与IFT(intraflagellar transport)依赖的动力蛋白臂组装的调节[14,16]。CCDC151-depleted斑马鱼的前肾和Kupffer’s囊的运动纤毛功能存在缺陷,并显示肾囊肿和随机左右不对称[13~14]。CCDC151在小鼠胚胎淋巴结中也有表达,且CCDC151缺陷可能导致小鼠纤毛不动或运动障碍,以及侧性缺陷[13]。Varsome数据库(https://varsome.com)查询结果显示,目前CCDC151基因上报道过的可能致病以及明确致病性变异约12个(表1),其中已报道的明确致病性变异均为无功能变异(c.325G>T,p.E109*[17];c.167delG,p.G56Dfs*26[12];c.925G>T,p.E309*[16,18];c.244+1G>A[19];c.583_595dup13[20];c.229_233delins32[21])。

表1 Varsome在线数据库查询CCDC151基因已收录变异情况Table 1 The included mutations of CCDC151 gene obtained from the Varsome online database
本研究通过先证者临床全外显子组测序技术,在中国人群中发现了CCDC151基因c.1059C>A(p.Tyr353*)纯合突变。该先证者为独生子,父母为近亲结婚,家系中仅先证者1名患者,临床表现为不育、内脏反位以及梗阻性无精子症,其中无精症是现有CCDC151基因变异中尚未描述过的表型。c.1059C>A变异位于CCDC151基因氨基酸高度保守区域,单个位点变异使原来编码酪氨酸的密码子改变为终止密码子。功能丧失是CCDC151基因(NM_145045.5)已知的疾病机制,该基因共13个外显子,而c.1059C>A变异位于第8号外显子,这种情况可导致NMD的发生,从而导致该基因编码蛋白质的过程提前终止。根据ACMG等指南,c.1059C>A突变可评估为致病性变异。总的来讲,本研究发现了1个Kartagener综合征家系的CCDC151基因新的无义突变,丰富了CCDC151基因突变谱,同时发现CCDC151基因纯合变异是导致患者不育及其梗阻性无精子症的可能遗传学病因,为后续辅助生殖治疗方案提供了重要指导。
纤毛和鞭毛运动障碍是诱发Kartagener综合征的根本原因[22],本研究中患者Kartagener综合征的诊断来自于外院,且患者不愿提供详细检查报告,仅为口述病情,故本研究可能有一定的局限性,对于梗阻性无精子症的诊断,缺乏睾丸体积、性激素、CT以及病理数据等梗阻性无精症诊断标准的结果描述,导致不能分析更详细的基因型-表现型关系。联合诊断有助于更详细的PCD诊断,并可能有助于诊断病理分型。
