Syntheses, Crystal Structures, Anticancer Activities and DNA-Binding Properties of the Dibutyltin Complexes Based on Benzoin Aroyl Hydrazone①
2022-03-12JIANGWuJiuTANYuJunMANJiTiTANYuXing
JIANG Wu-Jiu TAN Yu-Jun MAN Ji-Ti TAN Yu-Xing②
a (Key Laboratory of Functional Metal-organic Compounds of Hunan Province, Key Laboratory of Functional Organometallic Materials, University of Hunan Province, Hunan Provincial Engineering Research Center for Monitoring and Treatment of Heavy Metals Pollution in the Upper Reaches of XiangJiang River, College of Chemistry and Materials Science,Hengyang Normal University, Hengyang, Hunan 421008, China)
b (Emergency Department, Xiangya Hospital, Central South University, Changsha 410008, China)
ABSTRACT Dibutyltin benzoin benzoyl hydrazone complex I [C6H5(O)C=N-N=C(Ph)CH(Ph)O]2[(n-Bu)2Sn]2 and dibutyltin benzoin salicyl hydrazone complex II [2-(OH)-C6H4(O)C=N-N=C(Ph)CH(Ph)O]2[(n-Bu)2Sn]2 were synthesized and characterized by IR, 1H, 13C and 119Sn NMR spectra, HRMS, elemental analysis and thermal stability analysis, and the crystal structures were determined by X-ray diffraction. The crystal of complex I belongs to monoclinic system, space group P21/n with a = 11.1942(8), b = 10.4754(7), c = 23.6700(17) Å, β =101.529(2)°, Z = 2, V = 2719.6(3) Å3, Dc = 1.371 Mg·m-3, μ(MoKα) = 0.966 mm-1, F(000) = 1152, R = 0.0458 and wR = 0.1312. The crystal of complex II is of triclinic system, space group Pwith a = 9.9960(4), b =11.2466(4), c = 14.0509(5) Å, α = 69.4190(10)°, β = 70.0600(10)°, γ = 81.8090(10)°, Z = 1, V = 1389.69(9) Å3,Dc = 1.380 Mg·m-3, μ(MoKα) = 0.950 mm-1, F(000) = 592, R = 0.0284 and wR = 0.0690. In vitro antitumor activities of both complexes were evaluated by CCK8 method against three human cancer cell lines (MCF7,NCI-H460 and HepG2), and complex II exhibited better antitumor activity than I. The interaction between complexes and calf thymus DNA was studied by UV-vis, fluorescence spectroscopy and viscosity measurements.
Keywords: dibutyltin, complex, synthesis, crystal structure, DNA;
1 INTRODUCTION
Since the discovery of the first efficacious anticancer metallodrugs cisplatin in 1965 by Rosenberg[1], metal complexes with anti-cancer activity began to attract people.During the last few years, it is noticeable that organotin compounds occupied an important place in cancer chemotherapy reports[2-5]because of their cytotoxic effects,ability to bind with DNA, anti-proliferating nature, and apoptotic-inducing nature. Although there are many reports on the biological activity and structures of different organotin compounds[6-8], it is difficult for scientists to accurately predict the biological activity of drugs based on the chemical structures of drugs in advance, so a large number of potentially biologically activity molecules are synthesized and exploring the structure-activity relationship is academically helpful to lay the foundation for the development of new organometallic anti-cancer drugs.
The structure of the compound determines its properties, so the ligand plays an important role in geometry and affects the biological activities[9-11]. Therefore, we designed to use acylhydrazone as a ligand, which has peptide bonds and multiple sites, and plays a strong role in regulating the coordination of metals. In this paper, we have synthesized two dibutyltin complexes based on benzoin aroylhydrazone,and their structures were characterized. The inhibitory activities of the complex on cancer cells in vitro were tested,and the interactions with DNA were studied, which provides an important theoretical basis for the development of new metal antitumor drugs.
2 EXPERIMENTAL
2. 1 Instruments and reagents
Infrared spectrum (KBr) was recorded by the Prestige-21 infrared spectrometer (Japan Shimadzu, 4000~400 cm-1).1H,13C and119Sn NMR spectra were measured with a Bruker AVANCE-500 NMR spectrometer. The elemental analysis was determined by PE-2400(II) elemental analyzer.Crystallographic data of the complexes were collected on a Bruker SMART APEX II CCD diffractometer. HRMS was measured with the Thermo Scientific LTQ Orbitrap XL (ESI source). Melting points were determined using an X4 digital microscopic melting point apparatus without correction(Beijing Tektronix Instrument Co. Ltd.). Thermogravimetric analyses (TGA) were recorded on a NETZSCH TG 209 F3 instrument at a heating rate of 20 ℃·min-1from 40 to 800 ℃under air. The UV spectra were determined with the UV-2550 spectrophotometer (Shimazu). Fluorescence spectra were obtained with a Hitachi F-7000 spectrophotometer with quartz cuvette (path length = 1 cm).
Benzoin benzoyl hydrazone and benzoin salicyl hydrazone ligand were prepared according to the literatures[12]. Ethidium bromide (EB), calf thymus DNA and tris(hydroxymethyl)methyl aminomethane (tris) were from Sigma-Aldrich LLC. Cisplatin was from QiLu pharmaceutical CO., Ltd.Dibutyltin oxide was from Alfa Aesar (China) Chemical Co.,Ltd. Other chemicals were from Sinopharm Chemical Reagent Co., Ltd. All reagents of analytical grade were obtained from commercial sources and used without further purification. The human breast cancer cells (MCF-7, ATCC No: HTB-22), human lung cancer cells (NCI-H460, ATCC No: HTB-177) and human liver cancer cells (HepG2, ATCC No: HB-8065) were obtained from American Tissue Culture Collection (ATCC). Culture media RPMI-1640 (10.0%) were purchased from USA GIBICO. Ultrapure water (18.2 MΩ·cm)obtained from a Milli-Q water purification system (Millipore Co., USA) was used in all experiments. Tris-HCl (0.01 mol·L-1) buffer solution was prepared by a certain amount of tris(hydroxymethyl)methyl aminomethane dissolved in ultrapure water before using, and the pH of the solution was adjusted to 7.40 with hydrochloric acid solution (0.1 mol·L-1).The purity of CT-DNA was determined by comparing the absorbance at 260 and 280 nm (A260/A280= 1.8~1.9/1). The concentration of CT-DNA was calculated by measuring the absorbance at 260 nm (ε260= 6600 L·mol-1·cm-1). The reserve solution was stored at 4 ℃. The ethidium bromide solution was prepared by a certain amount of ethidium bromide solid dissolved in tris-HCl (0.01 mol·L-1) buffer solution.
2. 2 Syntheses of the complexes
A mixture of dibutyltin oxide (1.0 mmol), benzoin benzoyl hydrazone ligand (1.0 mmol) and CH3OH (20.0 mL) was added in a round-bottomed flask (50.0 mL), and refluxed with stirring for 6.0 h. Then the mixture was cooled and filtered. The complex crystals were obtained by controlling solvent evaporation. Complex I was yellow crystal. Yield:86%. m.p.: 131~133 ℃. Anal. Calcd. (C58H68N4O4Sn2): C,62.05; H, 6.11; N, 4.99%. Found: C, 62.11; H, 6.07; N,4.91%. FT-IR (KBr, cm-1): 3086, 3053, 3028ν(=C-H), 2954,2922ν(-C-H), 1606ν(C=N), 686ν(Sn-O), 588ν(Sn-O-Sn),530ν(Sn-N), 430ν(Sn-C).1H NMR (500 MHz, CDCl3,δ/ppm): 8.13~8.15 (d,J= 7.4 Hz, 2H), 7.91~7.93 (m, 2H),7.46~7.49 (m, 1H), 7.38~7.43 (m, 5H), 7.14~7.23 (m, 5H),6.25 (s, 1H), 1.62~1.71 (m, 4H), 1.47~1.51 (m, 4H),1.34~1.40 (m, 4H), 0.85~0.90 (m, 6H).13C NMR (126 MHz,CDCl3,δ/ppm): 171.76, 165.94, 143.59, 133.84, 131.67,131.27, 130.71, 129.73, 128.60, 128.17, 128.10, 128.06,127.47, 77.78, 27.14, 27.06, 26.65, 26.56, 21.30, 20.63, 13.30,13.56.119Sn NMR (187 MHz, CDCl3,δ/ppm): -108.71.HRMS (ESI)m/zcalcd. for C58H68N4O4Sn2Na+[M+Na]+1145.31708, found 1145.31689.
Complex II was prepared in a similar procedure (Fig. 1) as I by benzoin benzoyl hydrazone ligand (1.0 mmol) in place of benzoin salicyl hydrazone ligand. The product was colorless crystal with the yield of 82%. m.p.: 164~166 ℃.Anal. Calcd. (C58H68N4O6Sn2): C, 60.33; H, 5.94; N, 4.85%.Found: C, 60.34; H, 5.98; N, 4.92%. FT-IR (KBr, cm-1):3458ν(-O-H), 3059, 3030ν(=C-H), 2954, 2922ν(C-H),1629ν(C=N), 671ν(Sn-O), 594ν(Sn-O-Sn), 501ν(Sn-N),459ν(Sn-C).1H NMR (500 MHz, CDCl3,δ/ppm): 12.01(s,1H), 7.99 (d,J= 8.4 Hz, 1H), 7.48-7.49 (m, 2H), 7.40-7.42(m, 3H), 7.34 (t,J= 7.7 Hz, 1H), 7.22~7.25 (m, 2H),7.17~7.19 (m, 3H), 6.89 (t,J= 7.7 Hz, 2H), 6.12 (s, 1H),1.65~1.73 (m, 4H), 1.49~1.60 (m, 4H), 1.36~1.42 (m, 4H),0.91 (t,J= 7.3 Hz, 4H).13C NMR (126 MHz, CDCl3,δ/ppm):172.59, 167.62, 160.00, 142.29, 133.47, 131.93, 130.92,129.57, 128.67, 128.62, 127.94, 127.82, 127.38, 118.73,117.08, 115.98, 78.29, 27.05, 27.01, 26.65, 26.52, 22.19,21.52, 13.61, 13.57.119Sn NMR (187 MHz, CDCl3,δ/ppm):-125.56. HRMS (ESI)m/zcalcd. for C58H68N4O6Sn2Na+[M+Na]+1177.30691, found 1177.30676.
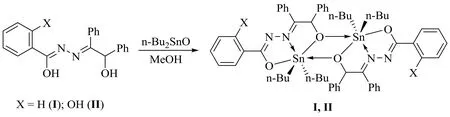
Fig. 1. Syntheses of the complexes
2. 3 Crystal structure determination
Suitable single crystals with dimensions of 0.41mm ×0.31mm × 0.16mm (I) and 0.48mm × 0.33mm × 0.23mm (II)were selected for data collection at 273(2) K on a Bruker SMART APEX II CCD diffractometer equipped with graphite-monochromated MoKαradiation (λ= 0.71073 Å)using aφ-ωmode. All the data were corrected byLpfactors and empirical absorbance. The structure was solved by direct methods. All non-hydrogen atoms were determined in successive difference Fourier synthesis, and hydrogen atoms were added according to theoretical models. All hydrogen and non-hydrogen atoms were refined by their isotropic and anisotropic thermal parameters through full-matrix leastsquares techniques. All calculations were completed by the SHELXTL-97[13]program. For complex I, a total of 37434 reflections were obtained in the range of 3.00<θ<26.00° with 5293 unique ones (Rint= 0.0314),S= 1.037, (Δρ)max= 1.012 and (Δρ)min= -0.897 e/Å3, max transmission was 0.8569,min transmission was 0.6916, and the completeness was 99.4%. For complex II, a total of 28937 reflections were obtained in the range of 3.10<θ<27.52° with 6372 unique ones (Rint= 0.0241),S= 1.059, (Δρ)max= 0.563, (Δρ)min=-0.4518 e/Å3, max transmission was 0.8111, min transmission was 0.6595, and the completeness was 99.6%.The selected bond lengths and bond angles for I and II are listed in Table 1.
2. 4 In vitro anti-tumor activity assays
The synthesized complexes were dissolved in DMSO at a concentration of 50 mM as stock solution. To avoid DMSO toxicity, the concentration of DMSO was less than 0.1% (v/v)in all experiments. MCF7, NCI-H460 and HepG2 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and grown at 37 ℃ in a saturated humidified atmosphere in the presence of 5% CO2. Cell proliferation was assessed by CCK8 assay[14-16]. 100 μL of cells (5 × 104cells·mL-1) was incubated at 37 ℃, and 5%CO2was seeded into 96 well plates. Then the medium was replaced with the respective medium containing complexes at different concentrations and incubated for 72 h. 10 μL CCK8 solution was added to each well and incubated at 37 ℃ for additional 4 h. The optical density was detected with a microplate reader at 450 nm. Six concentrations (5 nM~50 μM) were set for the compounds and at least 3 parallels of every concentration were used. All experiments were repeated at least three times. The data were calculated using Graph Pad Prism version 7.0. The IC50was fitted using a non-linear regression model with a sigmoidal dose response.
2. 5 Interaction with DNA
In the fluorescence study, a mixture of CT-DNA (30 μM),EB (3 μM) and different concentration complexes I or II solution (0~80 μM) was placed in a 5 mL volumetric flask in tris-HCl (0.01 mol·L-1) buffer solution[15,16]. After 3 h, the fluorescence spectra were acquired at 25 ℃. The excitation wavelength was 258 nm, and the emission wavelength is shown in the spectrum. The slit scanning width of emission and excitation is 5.0 nm. Finally, the quenching constant (Ksv)values of I or II were determined by using the Stern-Volmer equation[17].
In the UV-visible absorption spectrometry study, the investigation of the possible binding modes of complex to DNA and the calculation of the corresponding DNAbinding constants (Kb) were carried out by UV-vis spectroscopy. UV-visible absorption spectrometry experiments were carried out with a constant concentration of complexes I or II (50 μM) while the concentration of CT-DNA (0~50 μM) in tris-buffer was varied. The intrinsic binding constants (Kb) were calculated according the following Wolfe-Shimmer equation[18]:

wherecDNAis the concentration of CT-DNA,εAthe observed extinction coefficient at arbitrary DNA concentration,εFthe extinction coefficient of the free complex, andεBthe extinction coefficient of the complex when fully combining to CT-DNA. The DNA-binding constantsKbgiven by the ratio of the slope to the intercept were determined by the Wolfe-Shimmer equation and the plotscDNA/(εA-εF) versuscDNA.
The viscosity measurements were carried out in the Ubbelohde viscometer in a water bath with 25.00 ±0.02 °C. Viscosity experiments were performed by fixed DNA concentration (50 μM) and increasing the concentration of complex I or II (0~50 μM). The flow time of sample was measured with a digital stopwatch.The values of viscosity were calculated from the flow times of DNA containing solutions corrected for the flow time of buffer alone (t0),η= (t-t0)[19]. Data were presented as (η/η0)1/3versus (ccomplex/cDNA), whereηis the viscosity of DNA in the presence of the complex,η0the viscosity of DNA alone,ccomplexthe concentration of the complex andcDNAthe concentration of DNA.
3 RESULTS AND DISCUSSION
3. 1 Spectral analyses
In FT-IR spectra of I, the characteristic peaks ofν(Sn-O),ν(Sn-O-Sn),ν(Sn-N) andν(Sn-C) occur at 686, 588, 530 and 430 cm-1, and the same characteristic peaks of II are located at 671, 594, 501 and 459 cm-1[20], respectively, which indicate similar structures for both complexes. The above results are confirmed by single-crystal X-ray diffraction analysis.
In the1H NMR spectrum, the integral area ratio of each peak is consistent with the number of protons in each group of the expected structure[21]. The hydrogen proton absorption peaks of the aryl ring of complexes I and II are observed at 7.14~8.15 and 6.89~7.99 ppm in the low field, respectively.The -CH(Ph)O- protons in I and II appear at 6.25 and 6.12 ppm, respectively. The -OH protons in benzoin salicyl hydrazone ligand of II appear at 12.01 ppm. Methyl and methylene on the butyl group peak are observed at the high field position.
The13C NMR spectra show carbon atoms from carboxyls of I and II have the same peak positions, and those of other groups are consistent with the number of structural carbon atoms theoretically speculated. It can be inferred from the positions of hydrogen proton and carbon peaks of both complexes that I and II have similar structures, which is consistent with the results of X-ray single-crystal diffraction.
The119Sn NMR spectrum indicates Sn-core peaks of I and II are a single peak with -108.71 and -125.56 ppm,respecttively, showing the existence of a single organotin complex in both complexes.
In HRMS spectra, mass spectral peaks of complex I appear atm/z1145.31689, which can be attributed to the absorption peaks of [M+Na]+. Complex II is found atm/z1177.30676 due to the absorption peaks of [M+Na]+.
3. 2 Structure description
The molecular structures of I and II are shown in Fig. 2. It can be seen from Fig. 2 that complexes I and II have similar molecular structures. They are centrosymmetric dimer distannoxane, with a Sn2O2four-membered ring in the middle of the molecule. The center of the ring is the symcenter of the molecule. In the four-membered Sn2O2ring, the sum of the interior angles is 360°, indicating this ring is planar. Each oxygen atom O(2) bridges the two tin atoms in an antisymmetric unit, with the Sn(1)-O(2) bond belonging to the normal Sn-O covalent bond: the Sn(1)-O(2)ibond distances (2.581 Å I, 2.849 Å II) are greater than the covalent radii of Sn and O (2.13 Å), while much less than the sum of the van der Waals radii of 3.69 Å.
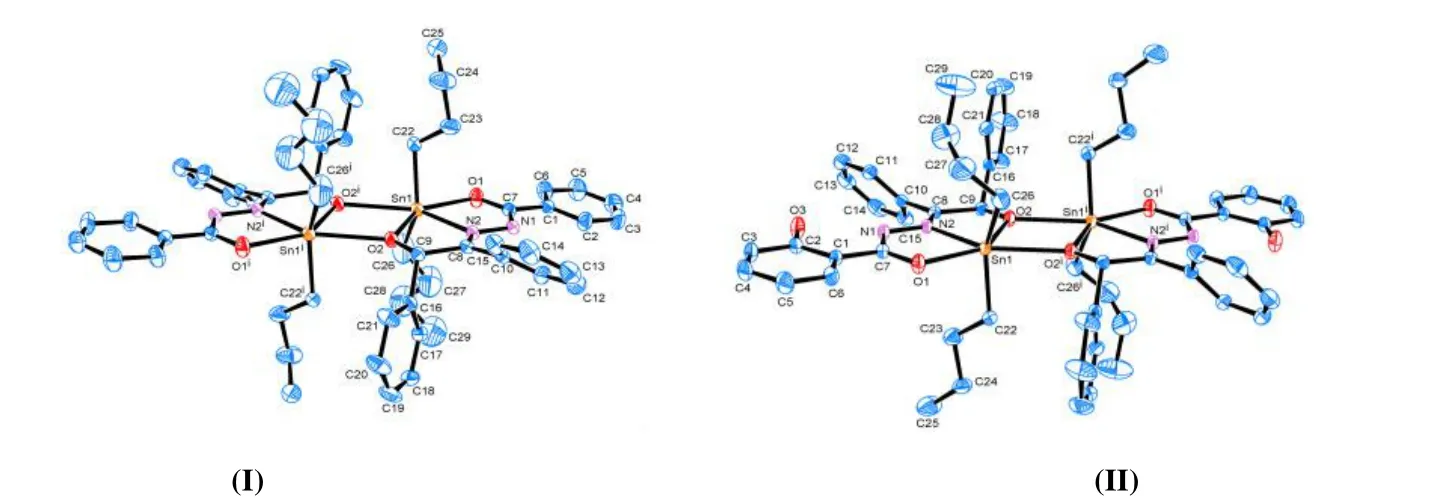
Fig. 2. Molecular structures of I and II. Symmetry codes for (I): i 1-x, 1-y, 1-z; (II): i -x+1, -y+1, -z
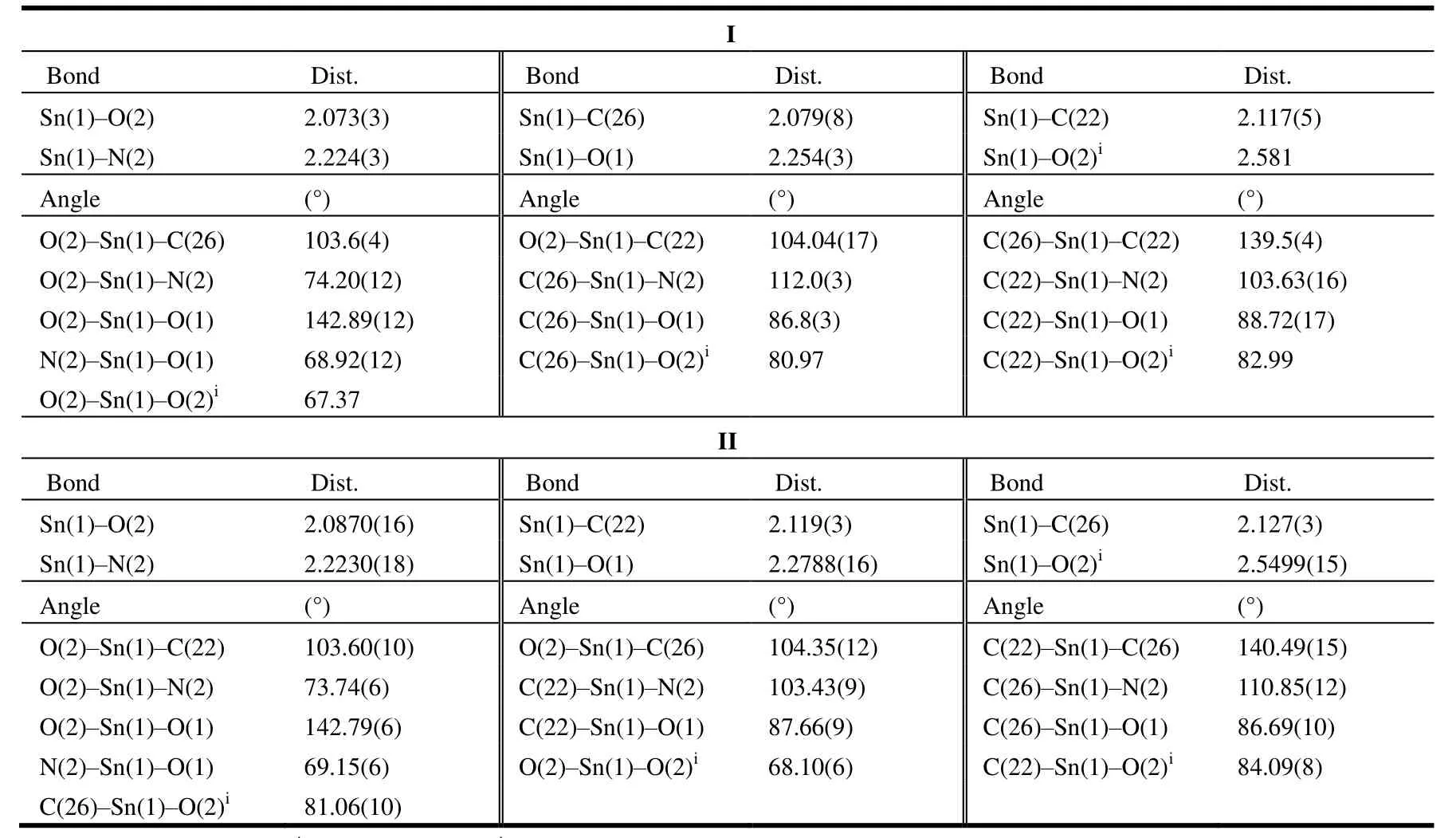
Table 1. Selected Bond Lengths (Å) and Bond Angles (°) for I and II
Take complex I as an example, the asymmetric unit comprises one benzoin benzoyl hydrazone ligand and a dibutyltin. The central tin atom is six-coordinated by two oxygen (O(1) and O(2)) and one nitrogen (N(2)) atoms from the ligand, two carbon atoms (C(22) and C(26)) from the butyl group, and one oxygen atom (O(2)i) from the other ligand. The axial positions are occupied by two methylene carbon atoms (C(22) and C(26)) from the butyl group, and the C(22)-Sn(1)-C(26) angle is 139.5(4)°, which obviously deviates from 180°. The equatorial positions are occupied by O(1), O(2), N(2) and O(2)i, and the bond lengths and angles between the five atoms and the tin atom are different, so in I the central tin atom is six-coordinated in a distorted octahedral geometry, and similar coordination mode is for II.
3. 3 Thermal stability
Thermal stabilities of both complexes were carried out using a NETZSCH TG 209 F3 thermogravimetric analyzer from 40 to 800 ℃ at a rate of 20 ℃·min-1under an air atmosphere at a flowing rate of 20.0 mL·min-1. As shown in Fig. 3, complexes I and II have no weight loss at 40 to 180 ℃. This shows that there is not solvent molecule involved in coordination in complex, which is consistent with X-ray single-crystal diffraction data. In the next stages, both complexes suffer complete decomposition until 800 ℃,corresponding to the removal of ligand and the dibutyl group attached to the tin atom. The remaining weight (27.40% (I)and 26.26% (II)) indicates the final products are SnO2(26.85% (I) and 26.11% (II)). In summary, I and II are rather stable up to about 200 and 180 ℃, respectively.

Fig. 3. TG-DTG curves for I and II
3. 4 Anticancer activity
The in vitro antitumor activities of complexes I and II were evaluated by CCK8 assay against MCF-7, HepG2 and NCI-H460 cell lines, using cisplatin as the positive control,with the results shown in Table 2. The anticancer activity of the ligand is relatively poor and there is no selectivity against MCF-7, HepG2 and NCI-H460 cell lines, but complexes I and II have obvious inhibitory effects on these three cancer cells, and they are better than cisplatin. Among three cancer cells, complexes I and II are also the most sensitive to HepG2, withIC50of 1.43 ± 0.72 μM and 0.73 ± 0.21 μM,respectively. In a word, complex II has slightly stronger inhibitory effect than I. From the test results, we can see significant statistical difference between complexes (or ligands) and cisplatin (P< 0.05). The molecular structure difference between I and II is only the substituents attached to the benzene ring on ligand by crystal structure analysis.Thereby, it can be preliminarily inferred that the substituents on the ligand have only a slight effect on the anti-cancer activity, which may be related to the synergistic effect of the ligand and organotin.
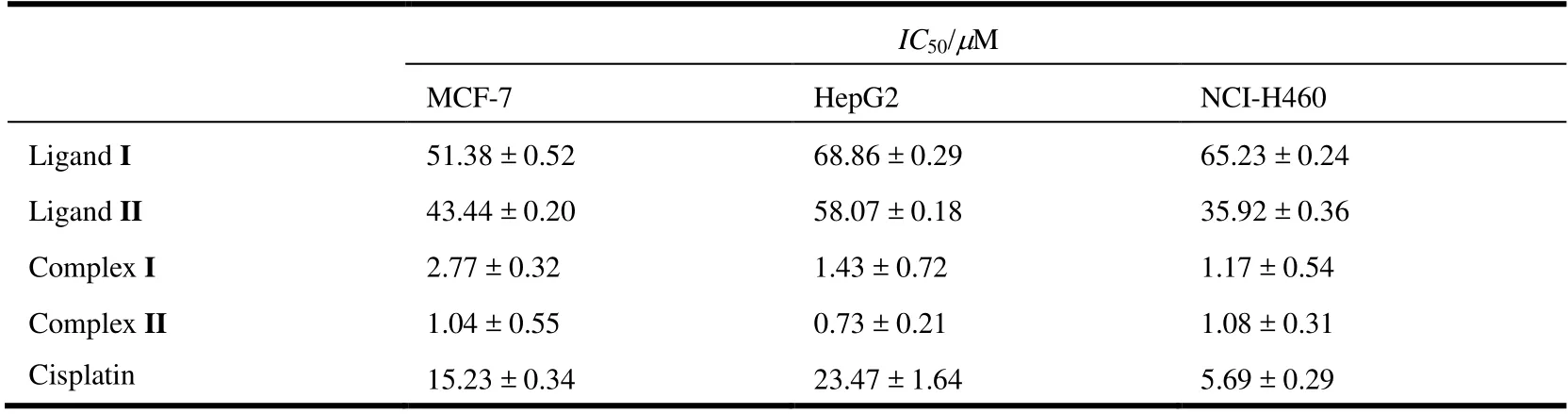
Table 2. Inhibition Action of the Complexes to Cancer Cell in vitro
3. 5 Interaction with DNA
Fig. 4 shows the effects of I and II on the fluorescent spectra of EB-DNA system with different concentrations.With increasing the concentration of complex, the fluorescence of EB-DNA complex system is quenched. This shows that complexes I and II interact with the EB-DNA system. The reasons may be as follows: (1) The complex will compete with EB for binding to the site on CT-DNA. The more complexes are added, the more EB originally bound to CT-DNA will be replaced by the complexes, which will reduce the fluorescence intensity of the EB-DNA system. (2)The complex and CT-DNA will compete to bind to EB,resulting in a decrease in the concentration of EB originally bound to CT-DNA, and it caused fluorescence quenching of EB-DNA. (3) The complex forms a new non-fluorescence compound with the EB-DNA system, that is, the DNA-EB-complex system, which leads to a decrease in the fluorescence intensity. In addition, the fluorescence spectrum of EB did not change significantly with the addition of complex I or II, indicating no interaction between the complexes and EB. And combined with the UV-visible absorption spectra, the binding between complexes I or II and CT-DNA is an embed mode. This basically eliminates reasons (2) and (3). In a word, the complex has a competitive effect with the DNA-EB system. To study quantificationally the binding capacity of complexes I or II and DNA[22,23], we employed the classical Stern-Volmer equationI0/I= 1 +KSVccomplexto obtain the quenching constantsKSVof complexes replacing EB and DNA with 1.0 × 105L·mol-1(I)and 2.5 × 105L·mol-1(II). The result showed that the binding strength of complex II is higher than that of I.
Fig. 5 shows the effects on UV-visible absorption spectra of I and II in the absence and presence of CT-DNA. The electronic absorption spectral data showed that when increasing the concentration of CT-DNA (0~80 μmol·L-1)added to complex I or II, both of the abovementioned absorption bands showed hypochromism and redshift (about 5 nm). The rate of hypochromism is 25% for I and 32% for II.According to the literatures[24,25], the DNA and the complex molecules are intercalation as probable mode of interaction,and the ultraviolet spectrum will have a significant hypochromism and accompany the red shift. The reason for the hypochromism is that the interaction between the complex and DNA causes the change of molecule conformation. And the change of the spectrum is related to the binding force. The stronger effect, the more obvious hypochromism. This phenomenon explains why the complex molecule inserts into the DNA. This preliminary result indicated that I or II intercalates into the DNA. TheKbvalues of complexes were calculated as 3.25 × 103L·mol-1(r2= 0.98)(I) and 4.98 × 103L·mol-1(r2= 0.99) (II) from the absorption spectra, and the high binding constant values reveal that the complex strongly interacts with DNA. It shows that complex I is stronger than II. The value is similar to those of complexes reported in the literature[26,27].
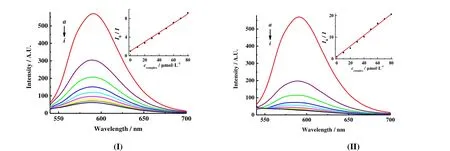
Fig. 4. Effects of II on the fluorescent spectra of EB-DNA system cCT-DNA = 30 μmol·L-1; cEB = 3 μmol·L-1; from a to i,ccomplex = 0, 10, 20, 30, 40, 50, 60, 70 and 80 μmol·L-1, respectively; inset: plot of I0/I vs. ccomplex; λex = 258 nm
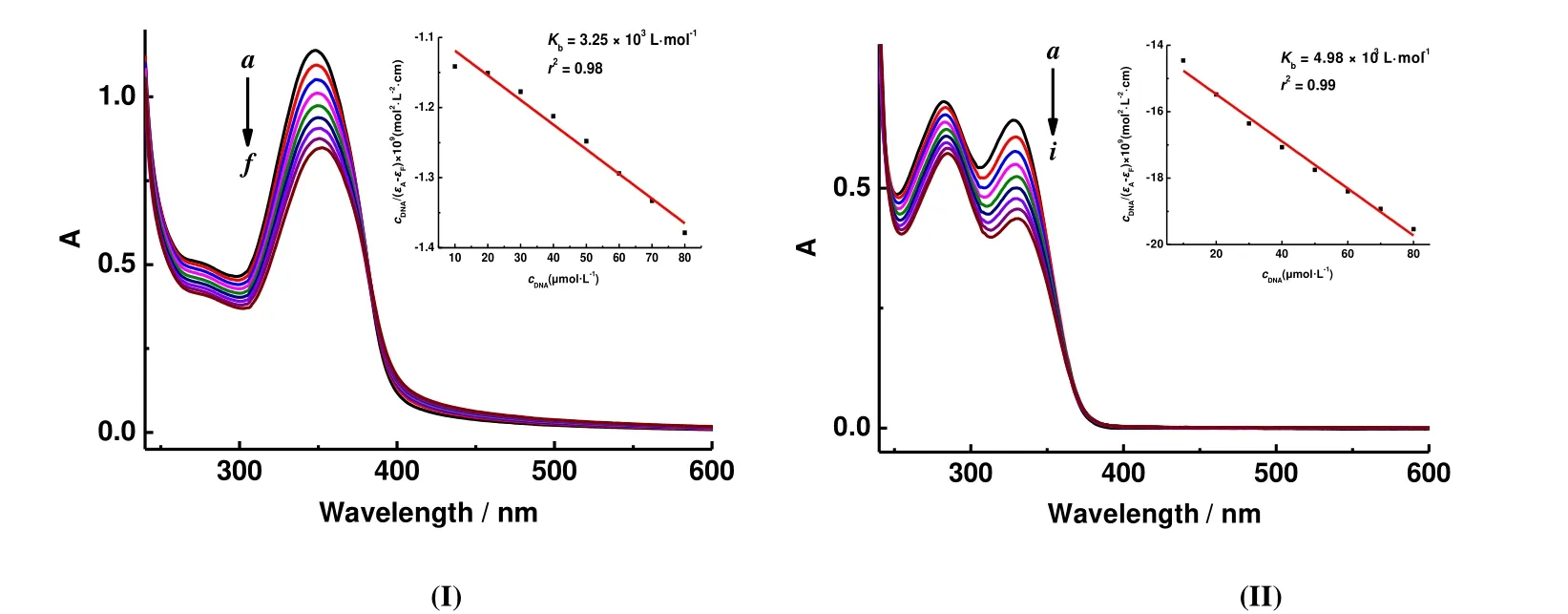
Fig. 5. UV-visible absorption spectra of II in tris-HCl buffer upon addition of CT-DNA (ccomplex = 50 μmol·L-1);from a to i, cDNA = 0, 10, 20, 30, 40, 50, 60, 70 and 80 μmol·L-1, respectively. The arrow shows the absorbance changes with increasing the DNA concentration. Inset: plot of cDNA/(εA - εF) vs. cDNA
The literatures[28,29]reported that a significant increase in the viscosity of DNA on the addition of a complex indicates the classical intercalative mode of binding to DNA. In contrast, if the interaction of the complex with DNA is caused by partial or non-classical insertion, the viscosity change of the DNA solution is not obvious or unchanged. As can be seen from Fig. 6, the relative viscosity of CT-DNA steadily increases with the concentration of the complex, and its trend is consistent with EB with the viscosity increase order to be EB > II >I. It shows that both the complex and EB interact with DNA in a similar mode. This observation can be explained by the fact that the complexes and DNA adopt a classical intercalation model, which requires that the DNA helix must be extended, resulting in an increase of DNA viscosity. The result further suggests an intercalating binding mode of the complexes with DNA and also parallels the above spectroscopic results.
4 CONCLUSION
Two dibutyltin (IV) complexes were synthesized.Complexes I and II are also a centrosymmetric dimer. II is stronger against MCF-7, HepG2 and NCI-H460 cell lines in vitro antitumour activity. The DNA-binding studies using UV-visible absorption spectroscopy showed that the intrinsic binding constant,Kb, was 3.25× 103L·mol-1(I) and 4.98 ×103L·mol-1(II). The fluorescent spectra showed that the Stern-Volmer quenching constant,KSV, was calculated as 1.0× 105L·mol-1(I) and 2.5 × 105L·mol-1(II). Therefore, both the complex and EB interact with DNA in a similar mode,thus proving that complexes and DNA adopt a classical intercalative binding, and the binding strength of complex II is higher than that of I.

Fig. 6. Effect of increasing amounts of the complexes and EB on the relative viscosity at 25.00 ± 0.02 °C
杂志排行

结构化学的其它文章
- Synthesis, Crystal Structure and Fungicidal Activity of 3,4-Dichloro-5-(6-chloro-9-(4-fluorobenzyl)-9H-purin-8-yl)isothiazole①
- Synthesis, Crystal Structure and Antifungal Activity of New Furan-1,3,4-oxadiazole Carboxamide Derivatives①
- Synthesis, Crystal Structure, Fungicidal Activities and Molecular Docking of Acyl Urea Derivatives Containing 2-Chloronicotine Motif①
- Molecular Design and Property Prediction of High Density 4-Nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate Derivatives as the Potential High Energy Explosives①
- A New Borate-phosphate Compound CsNa2Lu2(BO3)(PO4)2:Crystal Structure and Tb3+ Doped Luminescence①
- Temperature-controlled Structural Diversity of Two Cd(II)Coordination Polymers Based on the Dicarboxylate Ligand①