Molecular Design and Property Prediction of High Density 4-Nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate Derivatives as the Potential High Energy Explosives①
2022-03-12YANGJingPANGYuLIMinXinYANGGeFeiJIAJingXinMENGXingJunLIULiHuYANGXioChunGAOXioZhen
YANG Jing PANG Yu LI Min-Xin YANG Ge-Fei JIA Jing-Xin MENG Xing-Jun LIU Li-Hu YANG Xio-Chun GAO Xio-Zhen
a (Department of Chemistry, Tangshan Normal College, Tangshan 063000, China)
b (School of Chemical Engineering and Light Industry,Guangdong University of Technology, Guangzhou 510006, China)
c (Yangquan Municipal Key Laboratory of Quantum Manipulation,Shanxi Institute of Technology, Yangquan 045000, China)
ABSTRACT To search for potential energetic materials with large energy density and acceptable thermodynamics and kinetics stability, twelve derivatives of 4-nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate (named A~L) are designed and analyzed by using density functional theory (DFT) calculations at the B3LYP/6-311G**level of theory. The molecular heats of formation (HOF), electronic structures, impact sensitivity (H50), oxygen balance (OB) and density (ρ) are investigated by isodesmic reaction method and physicochemical formulas.Furthermore, the detonation velocity (D) and detonation pressure (P) are calculated to study the detonation performance by Kamlet-Jacobs (K-J) equation. These results show that new molecule J (H50 = 36.9 cm, ρ = 1.90 g/cm3, Q = 1912.46 cal/g, P = 37.82 GPa, D = 9.22 km/s, OB = 0.00), compound A (H50 = 27.9 cm, ρ = 1.93 g/cm3,Q = 1612.93 cal/g, P = 38.90 GPa, D = 9.19 km/s) and compound H (H50 = 37.3 cm, ρ = 1.97 g/cm3, Q = 1505.06 cal/g, P = 37.20 GPa, D = 9.01 km/s) present promising effects that are far better RDX and HMX as the high energy density materials. Our calculations can provide useful information for the molecular synthesis of novel high energy density materials.
Keywords: 4-nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate, energetic materials, density functional theory, explosive; DOI: 10.14102/j.cnki.0254-5861.2011-3256
1 INTRODUCTION
The value of harnessing the power of energetic materials(EMs) has been realized for quite some time, resulting in their pervasive use in different commercial processes[1-3]. Advancements in energetic materials have also been driven by a need to find more powerful, stable, and reliable materials for military devices. These traditional energetic compounds are explored such as trinitrotoluene (TNT), pentaerythritol tetranitrate (PETN), cyclotrimethylenetrinitramine (RDX),cyclotetramethylene tetranitramine (HMX), 1,1-diamino-2,2-dinitroethylene (FOX-7) and triaminotrinitrobenzene(TATB). They consist of organic C-H-N-O molecules which combine both fuel (C-H backbone) and oxidizer (nitro (NO2)or nitrate (NO3)) groups within a single mole cule[4]. Afterwards, much work has been focused on the derivatives of these traditional high energy density materials (HEDMs).Although such investigations provide some important results,the demands of high energy and insensitivity are quite often contradictory to each other, making the development of novel HEDMs a challenging problem. Thus, systemic molecular design for high-nitrogen compound is still needed to explore novel insensitive HEDMs.
As we all know that these compounds containing triazole ring, as an important class of high-energy density materials(HEDMs), have received vital attention both in military and civilian applications[5,6]. Recently, Yang research group conduct the study on synthesis of different neutral compounds consisting of 1,2,3-2H-triazole and 1,2,4-triazole rings carrying energetic moieties like amino, nitroimino, nitro as well as azo[7]. Unfortunately, their outstanding properties such as high density, high positive heat of formation (HOF) and excellent detonation properties seem to be contrary to the stability and sensitivity. Thus, in order to overcome this difficulty, one possible approach is to replace one hydrogen atom using different high-energy groups to design different derivatives. Thus, we choose excellent 4-nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate among these synthetic organic molecules as the initial material to develop energetic materials. So, in our work, twelve kinds of energetic groups(NO2, NH2, NHNH2, NHNO2, NNH2NO2, NNO2NO2,N3,ONO2, NNO2ONO2, OH, NF2and C(NO2)3) are introduced to this framework using computer simulation, and generate a series of high energy materials. Computational studies can provide understanding relationships between molecular structure and property, and make it possible to screen candidate compounds.
2 CALCULATION METHODS
Numerous researches have shown that the DFT-B3LYP method in combination with 6-311G** basis set can give accurate energies, molecular structures and physicochemical properties especially for the high energy density materials(HEDMs)[8-11]. Thus, the Gaussian 09 package[12]of theoretical chemistry was used in this paper under B3LYP/6-311G** level of theory to conduct our work. In our study, we used isodesmic reactions for calculating the HOF of the title molecules at 298 K as follows[8]:
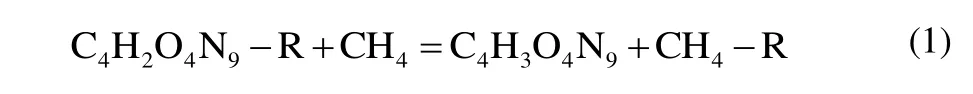
Where R is NO2, NH2, NHNH2, NHNO2, NNH2NO2,NNO2NO2,N3, ONO2, NNO2ONO2, OH, NF2, C(NO2)3(See Fig. 1). For isodesmic reaction, gas-phase HOF at 298 K can be written as the following formula:

Table1.Calculated Methodsforthe Valuesof N,,and Qof ExplosiveCaHbOcNd.isthe MolecularWeight ing/molandtheSolid PhaseHOF inkcal/mol

Table1.Calculated Methodsforthe Valuesof N,,and Qof ExplosiveCaHbOcNd.isthe MolecularWeight ing/molandtheSolid PhaseHOF inkcal/mol
Parameters Explosives components conditions c≥2a + b/2 2a + b/2>c≥b/2 b/2>cimages/BZ_139_423_2985_464_3026.png (b + 2c + 2d)/4M (b + 2c + 2d)/4M (b + d)/2Mimages/BZ_139_423_3040_468_3081.png4M/(b + 2c + 2d) (56d + 88c-8b)/(b + 2c + 2d) (2b + 28d + 32c)/(b + d)Q×10-3 (28.9b + 94.05a+ 0.239images/BZ_139_752_3159_831_3207.png(57.8c + 0.239)/M)/M[28.9b + 94.05(c/2-b/4)+ 0.239images/BZ_139_1214_3158_1296_3208.png]/M images/BZ_139_1806_3116_1896_3169.png


Fig. 1. All molecular structures of the title derivatives

3 RESULTS AND DISCUSSION
3. 1 Heats of formation
The basic structures are presented in Fig. 1. For the sake of discussion, all derivatives of 4-nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate are named A~L in sequence. Heat of formation (HOF) is a significant factor to reflect the energy content of a compound and molecular stability. High and positive HOF means high energy but less stability. The value of HOF (kJ/mol) is calculated based on formulas (2), (3) and(4), and the ultimate results are listed in Table 2. At the same time, Table 2 also lists the total energies (E0, a.u), zero-point energies (ZPE, a.u) and thermal corrections (HT, a.u) in the isodesmic reactions at the B3LYP/6-311G** level of theory.Besides, Table 2 presents the data of traditional energetic materials to compare our results in this section.
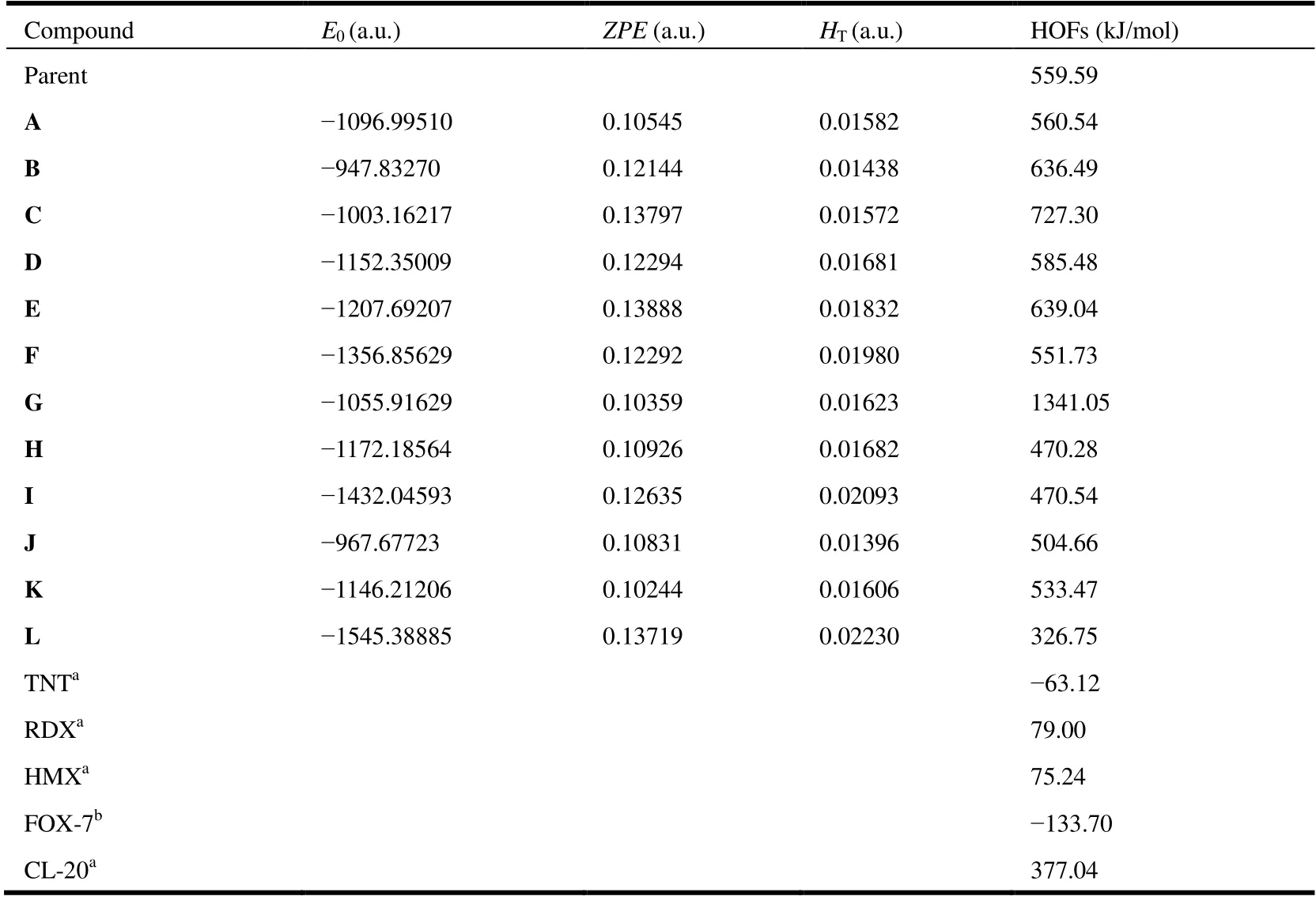
Table 2. Calculated Total Energies (E0, a.u.), Zero-point Energies (ZPE, a. u.), Thermal Corrections (HT, a. u.), and Solid Phase Heats of Formation (HOFs, Solid, kJ/mol) of Twelve New High Explosives Compared to Traditional Explosives
As shown in Table 2, all derivatives of the title compound have positive HOFs, which is one of the requirements for energetic materials[20]. It is worth noting that not only all derivatives exhibit excellent HOFs but also all the corresponding values (326.75~1341.05 kJ/mol) are higher compared with TNT (-63.12 kJ/mol), RDX (79.00 kJ/mol),HMX (75.24 kJ/mol) and FOX-7 (-133.70 kJ/mol)[15,21].These values, especially for compound G, meet most military and civilian requirements. The HOFs decrease to the lowest value (326.75 kJ/mol) when about three nitroso groups are present on the substitution site. The HOFs then obtain maximum (1341.05 kJ/mol) as additional -N3group is added to the original skeleton because abundant N-N bonds also have positive effect for increasing the HOFs value. When the substituent is NO2, NH2, NHNH2, NHNO2, NNH2NO2or N3,an increase HOF value of its substituted compounds is large when compared with the unsubstituted case. While the substituent is NNO2NO2, ONO2, NNO2ONO2, OH, NF2or C(NO2)3, the case is quite different. As expected, the introduction of nitrogen rich groups (NO2, NH2, NHNH2,NHNO2, NNH2NO2, N3) results in higher heats of formation than their parent (559.59 kJ/mol), which means these designed materials are promising to apply in the future. This change trend of HOF can be arranged in the sequence G > C > E > B > D > A > F > K > J > I > H > L.
3. 2 Molecular structures and electronic properties
Our method to design new high energy density materials on years of experience is coupled with interdisciplinary computational way in this field. There are three concrete standards that a newly-designed energetic explosive should meet: (i) stable five-membered heterocyclic rings obtained in our designed structures in order to stop abrupt dissociation/decomposition of these rings, (ii) a relatively-high number of nitrogen and oxygen atoms to guarantee lower oxygen balance (OB) and higher heat of formation (HOF), (iii)a combination of amino and nitro groups to improve stability to mechanical impact and thermal stability of the final materials. What’s more, one advantage of these compounds designed by us is that each new organic molecule contains two heterocycles, which greatly improves the stability of the materials.
The highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) are called frontier molecular orbitals (FMOs). The energy gap() between HOMO and LUMO reflects the chemical reactivity, kinetic stability and optical polarizability of the compounds. The molecular frontier orbital energy levels and their gaps are contained in Table 3. It is obvious to see from Table 3 that HOMO energies vary from -0.3027 to-0.3438 a.u. and LUMO from -0.1106 to -0.1602 a.u. for all new designed derivatives, and the frontier orbital energies differ from different compounds. Except for compound J, it is noteworthy that all the investigated molecules have higher energy gap than their parent and compound K with a NF2group has the maximum value. Thus, the contributions of the NF2group to the derivatives improve the stability of the compound and reduce their chemical activity. As shown in Table 3, the value of the investigated molecule J is less than the parent, indicating that the new designed molecule may be more sensitive than the parent. The differences predict that the chemical activity of the title compounds decreases in the following order: K > C > F > D > A > I > E > L > H > B >G > J. The low value ofmeans this electron transfer is easy from HOMO to LUMO. In other words, these molecules are more sensitive and chemically active and less stable.

Table 3. Frontier Orbital Energies and Their Differences of Title Compounds at the B3LYP/6-311G** Level of Theory
3. 3 Impact sensitivity
Apart from the energies and structures, the sensitivity of energetic materials is also a key study of keen interest to researchers in the research area of high energy density materials. Here, we use characteristic height (H50) to evaluate the sensitivity of molecules. Theoretical prediction of impact sensitivity for energetic materials has long been considered a difficult study, because the sensitivity of organic compounds is relevant to their decomposition kinetics and thermodynamics, which is very complicated. Actually, a calculation using crystal volume factors to assess impact sensitivities of nitramine energetic is proposed by Politzer and co-workers,which give acceptable accuracy[18]. So, the impact sensitivities of new organic derivatives were also computed by using model 1 forH50values. Besides, according to previous study,some researchers found that the compounds are stabilized by the delocalization of electronic charge. The impact sensitivity can be relevant to the degree of charge separation and the presence of strongly positive electrostatic potential maxima on the molecular surface[19]. Thus, in order to further discuss whether our system fits the relevant models, we also calculate theH50values of the other four models in this work. TheH50values of all five models are listed in Table 4. We can clearly see a big vary between the values of different models, which may be caused by the different influence factors considered by each formula. Because these designed molecules have no specific experimental values forH50, we only compare these values using four models with the experimental values of parent compound. For the experimental value ofH50, the parent is 24 cm; for the theoretical value, model 1 is 39.1,model 2 is 126.1 cm, model 3 is 60.98 cm, model 4 is 1.49 cm,and model 5 is 0.13 cm. These results support model 1 is significant in predicting impact sensitivities for our system,and the factors of other models may be minor. So next, we will focus on model 1 because its prediction results are more accurate than those of other models in our system.
In Table 4, these molecules haveH50values between 19.7 and 37.5 cm. Compound L with C(NO2)3group has the lowestH50, which means it is more sensitive, but compound B with the largest value is more stable. A NH2group appears in compound B, which may provide additional hydrogen bond interactions. Thereby, there is a significant increase inH50in this impact sensitivity. The impact sensitivityH50of these new molecules was nearly above 26 cm, and most of them were comparable with that of common explosives, RDX (26 cm)and HMX (29 cm)[22,23]. Besides, theH50values of all derivatives are higher than CL-20. The sensitivity increased with the number of oxygen atoms in different new groups. The phenomenon may result from oxides that have a strong power to attract electrons, thus reducing their impact sensitivity to some extent. However, owing to the complexity of assessing impact sensitivity, these views could be single rather than conclusive.
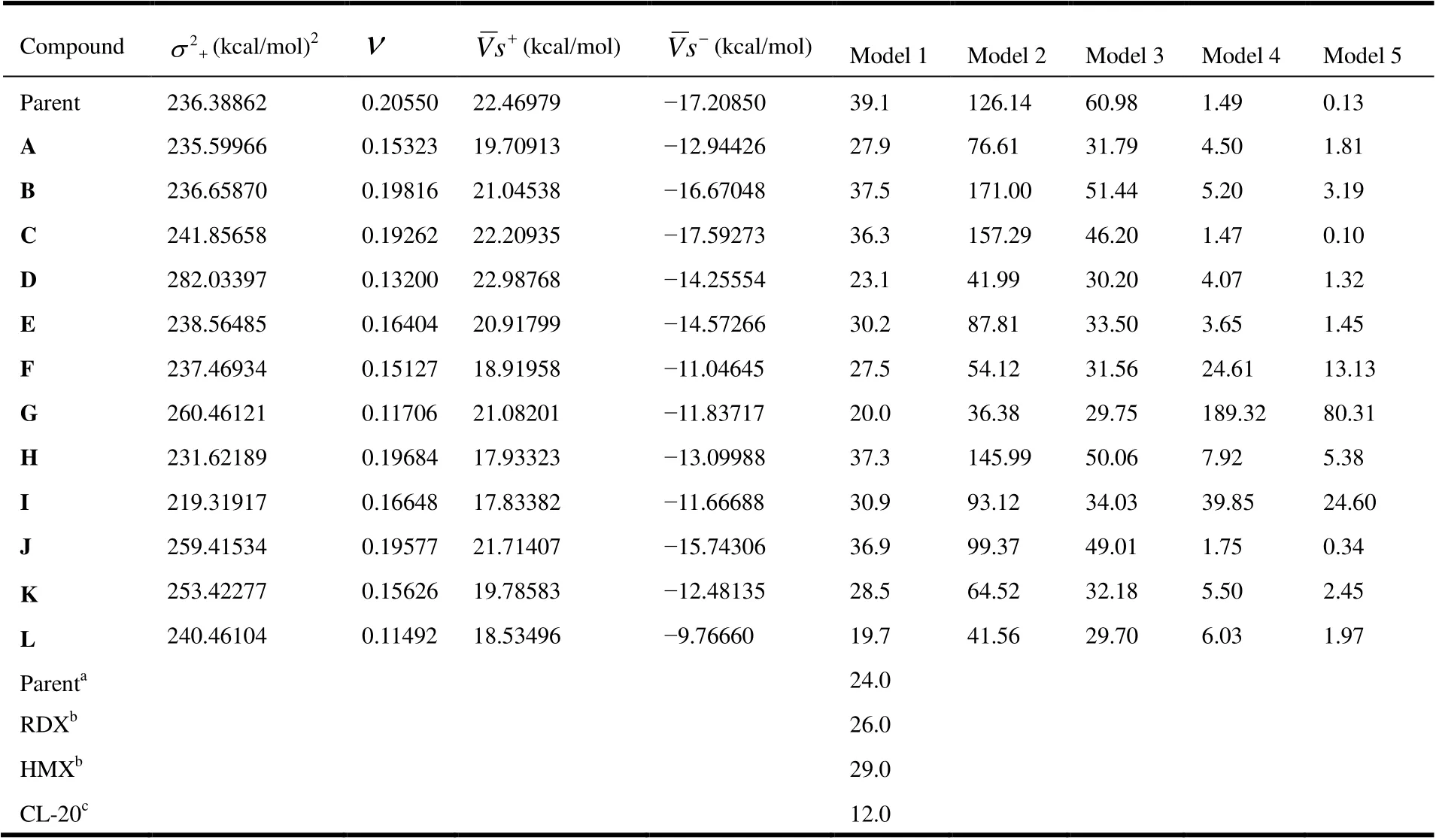
Table 4. Calculated Impact Sensitivity (H50) of the Investigated Molecules and RDX, HMX and CL-20
3. 4 Detonation performance
Detonation velocity (D) and detonation pressure (P) are two key explosive parameters for a high energy density material. These parameters can be calculated by the Kamlet Jacobs empirical equations on the basis of the theoretical density (ρ) and heat of detonation (Q) of the energetic materials. TheQandρvalues of synthetic molecules can be measured experimentally, but for some new organic materials,they are very difficult to obtain experimentally. Thus, these twovalues of designed new compounds have to be shown firstly to gain key parameters. Therefore, the calculated densities (ρ), heats of detonation (Q), detonation velocities (D)and detonation pressures (P) of the designed molecules are shown in Table 5. At the same time, the experimental detonation performances of traditional energetic materials are listed in Table 5 for an intuitive comparison.
From the table, we can see that values ofρ,Q,DandPare from 1.76 g/cm3(L) to 1.97 g/cm3(H), from 1011.45 cal/g (G)to 2083.31 cal/g (C), from 7.47 km/s (G) to 9.19 km/s (A),and from 24.28 GPa (G) to 38.9 GPa (A), respectively. By comparing theρvalue of parent and these new title compounds, we found that the density of most of the molecules is much higher than the density of parent. Besides,theρvalue of the parent is close to that of compound E.Further work was focused on improving the detonation performance of the title compounds by introducing energetic groups which resulted in the second-generation of agent defeat weapons. The introduction of NF2is a successful strategy, with the molecular density rising from 1.86 g/cm3(parent) to 1.98 g/cm3(K), which is better than that of TNT(1.64 g/cm3), RDX (1.80 g/cm3), HMX (1.90 g/cm3), and FOX-7 (1.89 g/cm3)[24]. As we know, a molecular density close to 2.0 g/cm3is desirable until now in the field of explosive. Thus, molecule K is glamorous from the viewpoint of density. As expected, the introduction of nitrogen rich groups leads to higher density than these common explosives have. Compounds A (1.93 g/cm3), F (1.92 g/cm3), H (1.97 g/cm3) and I (1.96 g/cm3) also show better density. After evaluating the physicochemical properties of the title compounds, including density and HOF, our attention has been turned to their detonation properties. As shown in Table 4, the calculated detonation velocity and detonation pressure of compound A are 9.19 km/s and 38.9 GPa, respectively,which are much superior to those of TATB (8.11 km/s and 32.4 GPa), TNT (6.95 km/s and 19.00 GPa) and RDX (8.75 km/s and 34.7 GPa)[24]. The detonation velocity and pressure of all compounds are also superior to those of TNT. But compound G (24.28 GPa) has lower detonation pressure than HMX (39.2 GPa), FOX-7 (35.9 GPa), and RDX (34.9 GPa)[24].
Furthermore, oxygen balance (OB) is also an important parameter for energetic materials to determine whether the compounds are oxygen-enriched or oxygen-poor. Here, for a compound with molecular formula CaHbOcNd, the oxygen balance can be represented as equation (9)[25]:

Table 5. Calculated Detonation Properties and Nitrogen Content of theTitle Compounds and Reference Compounds TNT, RDX, HMX, FOX-7 and CL-20
4 CONCLUSION
A family of new organic molecules of 4-nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate are designed. The molecular structures, electronic properties, heat of formation(HOF), impact sensitivity (H50), density (ρ), detonation velocity (D) and detonation pressure (P) of all new compounds are completely characterized by theoretical calculation at the B3LYP/6-311G** level of theory. Besides,oxygen balance (OB) and nitrogen content for all compounds are discussed in this paper. It is noteworthy that all title compounds have excellent HOFs and higherDandPvalues than TNT. Eight of them (A, C, D, E, F, H, I, J) have higherPvalues than RDX (34.70 GPa) and FOX-7 (34.00 GPa), and three of them (A, E, J) have higherDvalues than HMX (9.10 km/s). The good detonation performances of these compounds are caused by outstanding density. Surprisingly, more than half of these derivatives have higher density over 1.80 g/cm3.
Based on the above results, we can see that new molecule J(H50= 36.9 cm,ρ= 1.90 g/cm3,Q= 1912.46 cal/g,P= 37.82 GPa,D= 9.22 km/s, OB = 0.00), compound A (H50= 27.9 cm,ρ= 1.93 g/cm3,Q= 1612.93 cal/g,P= 38.90 GPa,D= 9.19 km/s) and compound H (H50= 37.3 cm,ρ=1.97 g/cm3,Q=1505.06 cal/g,P= 37.20 GPa,D= 9.01 km/s) can be considered as potential candidates in terms of the energetic material. Especially for compound J, the good balance of detonation performance and sensitivity, plus the environmental oxygen balance, contribute to its practical application as a promising primary explosive. It is a green and powerful alternative to the toxic and sensitive explosives.
杂志排行

结构化学的其它文章
- Synthesis, Crystal Structure and Fungicidal Activity of 3,4-Dichloro-5-(6-chloro-9-(4-fluorobenzyl)-9H-purin-8-yl)isothiazole①
- Synthesis, Crystal Structure and Antifungal Activity of New Furan-1,3,4-oxadiazole Carboxamide Derivatives①
- Syntheses, Crystal Structures, Anticancer Activities and DNA-Binding Properties of the Dibutyltin Complexes Based on Benzoin Aroyl Hydrazone①
- Synthesis, Crystal Structure, Fungicidal Activities and Molecular Docking of Acyl Urea Derivatives Containing 2-Chloronicotine Motif①
- A New Borate-phosphate Compound CsNa2Lu2(BO3)(PO4)2:Crystal Structure and Tb3+ Doped Luminescence①
- Temperature-controlled Structural Diversity of Two Cd(II)Coordination Polymers Based on the Dicarboxylate Ligand①