Synthesis, Crystal Structure and Fungicidal Activity of 3,4-Dichloro-5-(6-chloro-9-(4-fluorobenzyl)-9H-purin-8-yl)isothiazole①
2022-03-12WANGWeiBoLIUXiaoYuLIZhiXinyiGAOWeiLVYouLIKunGLUKHAREVATatianaTANGLiangFuFANZhiJin
WANG Wei-Bo LIU Xiao-Yu LI Zhi-Xinyi GAO Wei LV You LI Kun GLUKHAREVA Tatiana V.TANG Liang-Fu FAN Zhi-Jin②
a (State Key Laboratory of Elemento-organic Chemistry,College of Chemistry, Nankai University, Tianjin 300071, China)
b (Frontiers Science Center for New Organic Matter,College of Chemistry, Nankai University, Tianjin 300071, China)
c (School of Medicine, Nankai University, Tianjin 300071, China)
d (Ural Federal University Named after the First President of Russia B. N.Yeltsin, Yeltsin UrFU 620002, Ekaterinburg, Russia)
ABSTRACT 3,4-Dichloro-5-(6-chloro-9-(4-fluorobenzyl)-9H-purin-8-yl)isothiazole, a novel purine derivative,was synthesized by the cyclization of pyrimidine amine. Its structure was characterized by 1H NMR, 13C NMR, 19F NMR, H RMS and single-crystal X-ray diffraction. This compound 3 is crystallized from a mixed solvent of dichloromethane and n-hexane (1:2, v/v) for structural identification as monoclinic crystal system, space group P21/n with a = 11.66250(10), b = 8.21300(10), c = 17.77920(10) Å, V = 1676.34(3) Å3, Z = 4, Dc = 1.643 g/cm3,F(000) = 832.0 and μ = 6.301 mm-1. 22315 reflections were measured (8.43≤2θ≤158.10°), of which 3532 were unique (Rint = 0.0311) and used in all calculations. The final R = 0.0334 (I > 2σ(I)) and wR = 0.0842 (reflections).The title compound showed over 50% of growth inhibition against Botrytis cinereal, Cercospora arachidicola,Gibberella zeae, Rhizoctonia solani and Sclerotinia sclerotiorum at 50 mg/L, and its EC50 value against R. solani was 60.44 µmol/L, which was active at the same level as that of positive control diflumetorim with its EC50 value of 60.29 µmol/L and less active than YZK-C22 with its EC50 value of 12.32 µmol/L, respectively. Our studies discovered that the combination of bioactive substructures of isothiazole with purine could be an effective way to novel fungicide development.
Keywords: purine, synthesis, crystal structure, fungicidal activity;
1 INTRODUCTION
The purine (imidazo-[4,5-d]pyrimidine) skeleton is an important structural motif which plays an important role in different life related processes[1,2]. During the wide range of biological activities, purine structure is considered as a privileged scaffold in medicinal chemistry. Many drugs containing purine fragment have been developed for the treatment of asthma, inflammation, cancer and gastrointe stinal diseases[3-9]. In addition, some compounds with purine fragment, such as aureonuclemycin, are fungicides for plant disease control[10]. As active substructures, heterocyclic ring structures with both S and N atoms[11], especially 3,4-dichlo roisothiazole[12], showed good systemic acquired resistance and fungicidal activities in pesticide lead discovery.
The discovery of lead compounds is an important basis for novel pesticide development. Our group focused on agrichemical lead discovery, different pyrazole-thiazoles[13],pyrazole-aromatics[14], and thiadiazole derivatives[15]were found to show various degrees of fungicidal activity.YZK-C22 is a highly active fungicidal lead[16]. The research has shown that YZK-C22 does not act at traditional pesticide targets, but has a new potent target: pyruvate kinase (PK)[17].Based on the structure of the lead molecule YZK-C22 and its potent new target PK, 3,4-dichloro-5-(6-chloro-9-(4-fluoro benzyl)-9H-purin-8-yl)isothiazole was rationally designed(Fig. 1) and synthesized (Scheme 1) by the combination of bioactive substructures of purine and isothiazole, and its crystal chemical structure and fungicidal activity were evaluated here.
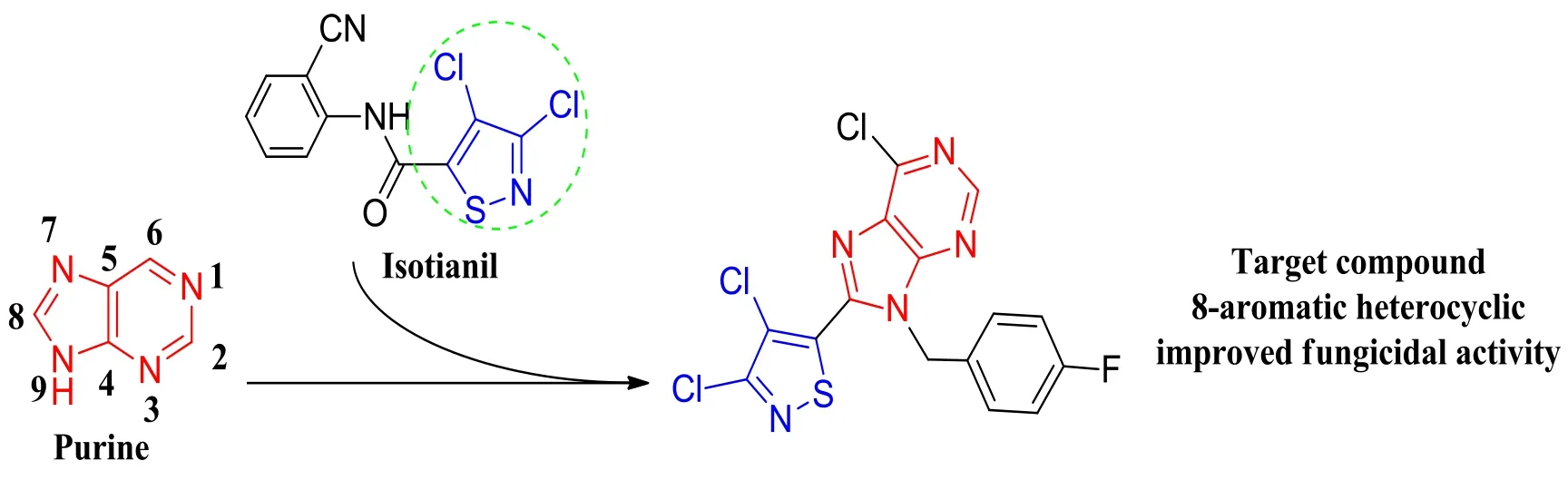
Fig. 1. Design of the target compound
2 EXPERIMENTAL
2. 1 Instruments and reagents
Melting point was measured on an X-4 Digital Type Melting Point Tester (Gongyi, China) and uncorrected.1H NMR spectra were recorded on a Bruker AV400 spectro meter (400 MHz) (Wisconsin, United States of America) and chemical shifts were reported in ppm.13C NMR spectra were recorded on a Bruker AV400 spectrometer (101 MHz)(Wisconsin, United States of America) with complete proton decoupling.19F NMR spectra were recorded on a Bruker AV400 spectrometer (101 MHz) (Wisconsin, United States of America) with complete proton decoupling. High-resolution mass spectra (HRMS) were recorded with an Agilent 6520 Q-TOF LC/MS instrument (Agilent Technologies Inc.State of California, United States of America). Crystal structure was determined on a Rigaku Xtalab P200 diffractometer. All of the solvents and materials were of reagent grade and purified as required.
2. 2 Synthetic procedure for the target compound
The procedure for the synthesis of compound 3 is shown in Scheme 1. As a key intermediate, pyrimidine amine 2 was synthesized according to the revision of the reported method[5]. Triethylamine (1.00 mL, 7.37 mmol) was added to a suspension of compound 1 (98% content) (1.00 g, 6.14 mmol) in ethanol (10 mL), followed by the addition of 4-fluorobenzylamine (0.75 mL, 6.45 mmol). Then the reaction mixture was stirred for 18 h at 80 °C. After the reactant was consumed, the reaction mixture was concentrated under reduced pressure to remove the solvent,and the intermediate 2 was obtained by purifying the crude residue using silica gel column chromatography with a mixture eluent of petroleum ether (60~90 °C fraction):ethyl acetate (2:1,v/v).
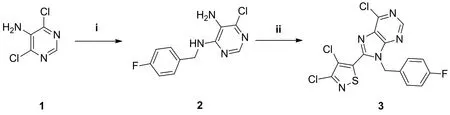
Reagents and conditions: (i) 4-fluorobenzylamine, EtOH, Et3N, 80 oC, 12 h (ii) 3,4-dichloroisothiazole-5-carbonyl chloride, NH4Cl, toluene, 100 oC, 2 h; POCl3, 100 oC, 12 h Scheme 1. Synthesis of the target compound 3
Analytical data for intermediate2. Yellow solid; yield,25%; m.p.: 221~223 °C.1H NMR (400 MHz, DMSO-d6)δ7.73 (s, 1H), 7.39~7.30 (m, 3H), 7.19~7.09 (m, 2H), 5.09 (s,2H), 4.60 (d,J= 5.6 Hz, 2H).13C NMR (101 MHz,DMSO-d6)δ161.2 (d,1JF-C= 242.2 Hz), 151.7, 145.5, 136.9,135.6 (d,4JF-C= 3.0 Hz), 129.3 (d,3JF-C= 8.2 Hz), 123.6,115.0 (d,2JF-C= 21.2 Hz), 43.4.19F NMR (101 MHz, CDCl3)δ-114.59. HRMS (ESI) m/z calcd. for C11H11ClFN4+(M+H)+:253.0651; found: 253.0649. Document[18]reported its yield of 84% with the m.p. of 240~242 °C.
Compound 3 was synthesized according to the revision of the reported method[19]. To a suspension of compound 2 (0.20 g, 0.79 mmol) in toluene, ammonium chloride (0.25 g, 4.74 mmol) and 3,4-dichloroisothiazole-5-carbonyl chloride (0.10 mL, 0.79 mmol) were added successively. The reaction mixture was heated at 100 °C for 2 h. After cooling the mixture to room temperature, phosphorus oxychloride (8.0 mL) was added. Then, the mixture was slowly heated to 100 °C again and kept for 12 h. After the reaction completed,the reaction mixture was slowly dropwise added to ice water.Then, the pH of the mixture was adjusted to 7~8 using ammonia water (25%~28%) carefully, and compound 3 in the mixture was extracted with ethyl acetate (15 mL × 3). The combined organic layers were washed with saturated sodium chloride solution (20 mL) for 3 times and dried over anhydrous sodium sulfate. After the solvent evaporation under reduced pressure, the residue of the target compound 3 was purified by silica gel column chromatography with a mixture of petroleum ether:ethyl acetate (5:1,v/v) as eluent.
Analytical data for compound3. Yellow solid; yield, 81%;m.p.: 133~134 °C.1H NMR (400 MHz, CDCl3)δ8.82 (s,1H), 6.99~6.92 (m, 2H), 6.92~6.85 (m, 2H), 5.53 (s, 2H).13C NMR (101 MHz, CDCl3)δ162.7 (d,1JF-C= 248.9 Hz), 153.1,152.9, 152.0, 149.4, 148.8, 143.7, 131.5, 130.0 (d,4JF-C= 3.2 Hz), 129.3 (d,3JF-C= 8.2 Hz), 124.2, 116.2 (d,2JF-C= 21.8 Hz), 47.2.19F NMR (101 MHz, CDCl3)δ-111.9. HRMS(ESI) m/z calcd. for C15H8Cl3FN5S+(M+H)+: 413.9545;found: 413.9549.
2. 3 Structure determination
The colorless crystal of the title compound 3 with dimensions of 0.18mm × 0.16mm × 0.13mm was cultured fromn-hexane/dichloromethane and selected for X-ray diffraction analysis. The data were collected on a Rigaku Xtalab P200 Single Crystal diffractometer equipped with mirror-monochromatic CuKαradiation (λ= 1.54184 Å) with anωscan mode at 294.15 K. In the range of 4.22≤θ≤79.05°, a total of 22315 reflections were collected with 3532 unique ones (Rint= 0.0311), of which 3238 were observed withI> 2σ(I) for refinements. Using Olex2[20], the structure was solved by the ShelXT[21]structure solution program using Intrinsic Phasing and refined with the ShelXL[22]refinement package using Least Squares minimization. All of the non-hydrogen atoms were located with successive difference Fourier syntheses. The hydrogen atoms were added according to theoretical models. The final full-matrix least-squares refinement converged atR= 0.0310,wR=0.0842 (w= 1/[σ2(Fo)2+ (0.0393P)2+ 0.5096P], whereP=(Fo2+ 2Fc2)/3),S= 1.075, (Δρ)max= 0.24, (Δρ)min= -0.25 e/Å3and (Δ/σ)max= 0.001.
2. 4 Fungicidal activity determination
The fungicidal activities of intermediate 2 and target compound 3 were evaluated at 50 mg/L according to the previously reported procedures[23-25]. Seven representative fungi,A. s:Alternaria solani;B. c:Botrytis cinerea;C. a:Cercospora arachidicola;G. z:Gibberella zeae;P. p:Physalospora piricola;R. s:Rhizoctonia solaniandS. s:Sclerotinia sclerotiorum, were tested. The commercially available pyrimidinamine fungicide diflumetorim and lead molecule YZK-C22 were selected as positive controls.Inhibitory rates (%) = (Dcontrol- Dtest)/(Dcontrol- 4) × 100,where Dcontrolwas the average diameter (mm) of mycelia in the absence of any compounds and Dtestwas the average diameter (mm) of mycelia treated with the test compound. All experiments were tested in triplicates. Data were presented as the mean ± standard deviation. EC50of the target compound 3 and corresponding positive controls againstR. solaniwere evaluated, too[16].
3 RESULTS AND DISCUSSION
As shown in Scheme 1, the target compound 3 was synthesized in good yield by cyclization of pyrimidine amine 2 with 3,4-dichloroisothiazole-5-carbonyl chloride. Its structure was characterized by1H NMR,13C NMR,19F NMR and HRMS.The crystal structure of compound 3, crystallizing from a mixed solvent of dichloromethane andn-hexane (1:2, v/v), is shown in Fig. 2.
The selected bond lengths, bond angels and torsional angels of compound 3 are shown in Tables 1 and 2. The bond lengths and angles of the isothiazole ring agreed well with the values reported[26]. Meanwhile, bond lengths and angles of the purine substructure appeared to be normal relative to the closely related compounds in literature[27]. The sum of C(4)-N(5)-C(9), C(8)-N(5)-H(9) and C(8)-N(5)-H(5)angles was 359.96°, indicating thesp2hybridization state of N(5) atom. The torsion angle of N(2)-C(5)-C(8)-N(4) is-178.75°, indicating that the whole purine was coplanar. The torsion angles of C(2)-C(3)-C(4)-N(2) and C(8)-N(5)-C(9)-C(10) are -67.7° and 121.03°, which means that both the isothiazole and benzene rings were nonplanar with the purine ring. As shown in Table 3, the intermolecular hydrogen bonds C(9)-HA(9)···F(1)i, C(9)-HA(9)···Cl(2)iiand C(9)-HB(9)···N(2)iiiwere found in compound 3, which lead to the position of benzene ring close to the isothiazole ring rather than the purine ring. These intermolecular hydrogen bonds stabilize the crystal packing (Fig. 3). In addition, the intermolecular C-H···πinteraction of C(12)-H(12)···C(15)iv(H(12)···C(15)iv2.676 Å) was also observed in the crystal packing of compound 3, which is two-dimensional. Noπ-πinteraction was observed due to the large distance between adjacent benzene ring and isothiazole ring or purine ring.
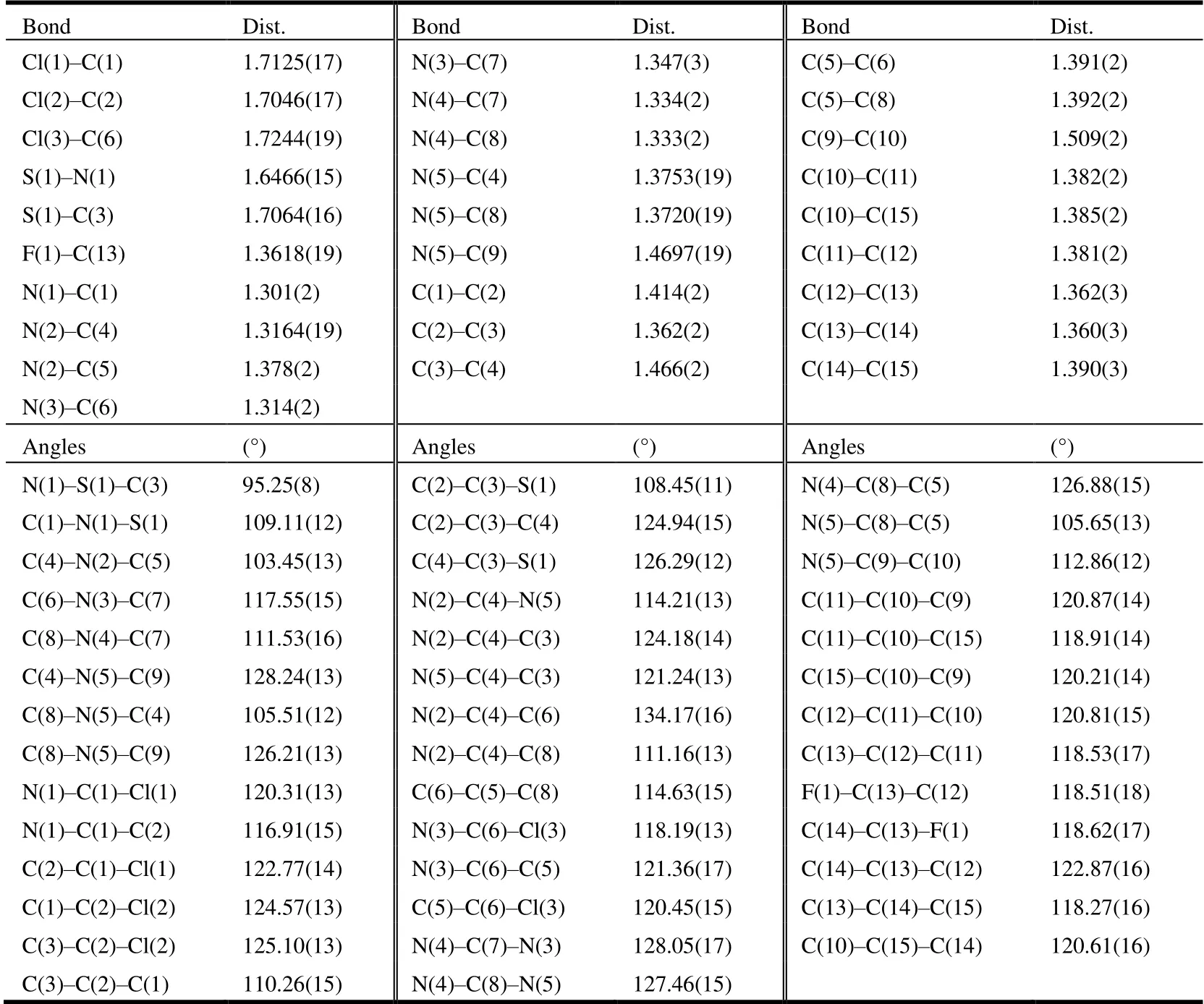
Table 1. Selected Bond Lengths (Å) and Bond Angles (°) for Compound 3
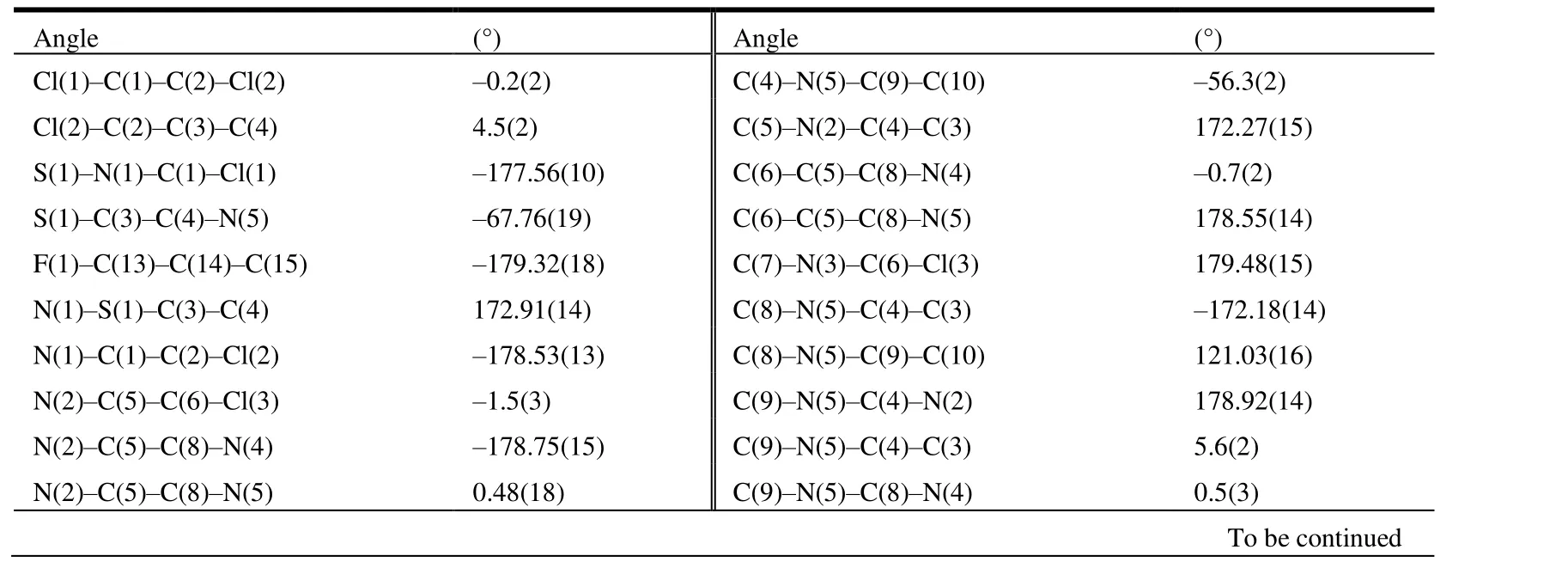
Table 2. Selected Torsional Angles (°) for Compound 3

N(5)-C(9)-C(10)-C(11) -49.3(2) C(9)-C(10)-C(11)-C(12) -178.15(16)N(5)-C(9)-C(10)-C(15) 131.96(16) C(9)-C(10)-C(15)-C(14) 177.84(17)C(2)-C(3)-C(4)-N(2) -67.7(2) C(11)-C(12)-C(13)-F(1) 179.01(18)C(2)-C(3)-C(4)-N(5) 104.98(18) C(13)-C(14)-C(15)-C(10) 0.6(3)C(4)-N(5)-C(8)-N(4) 178.30(15) C(15)-C(10)-C(11)-C(12) 0.6(3)
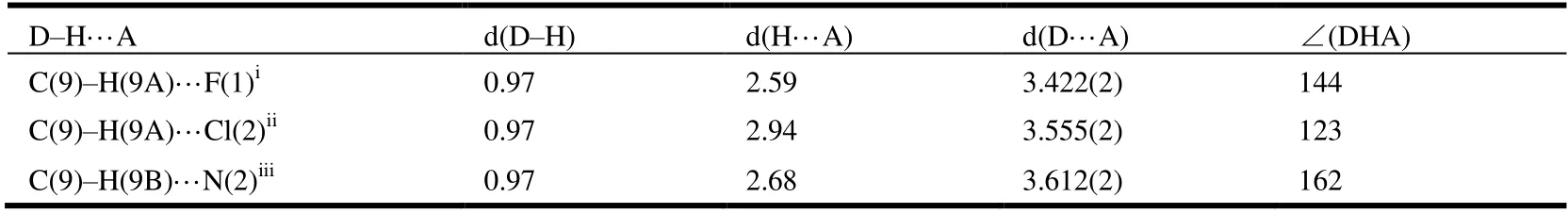
Table 3. Hydrogen Bond Lengths (Å) and Bond Angles (°) for Compound 3
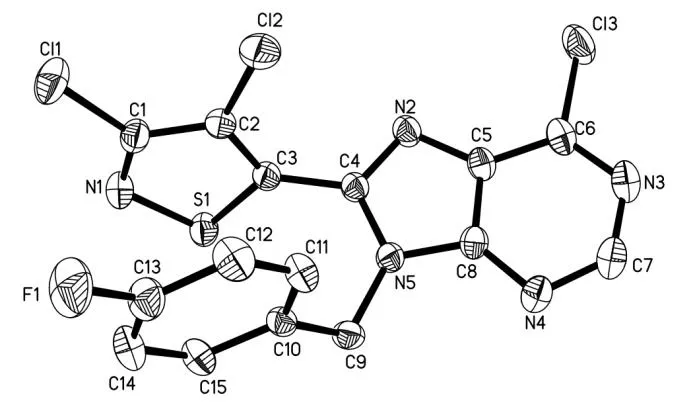
Fig. 2. X-ray crystal structure of compound 3
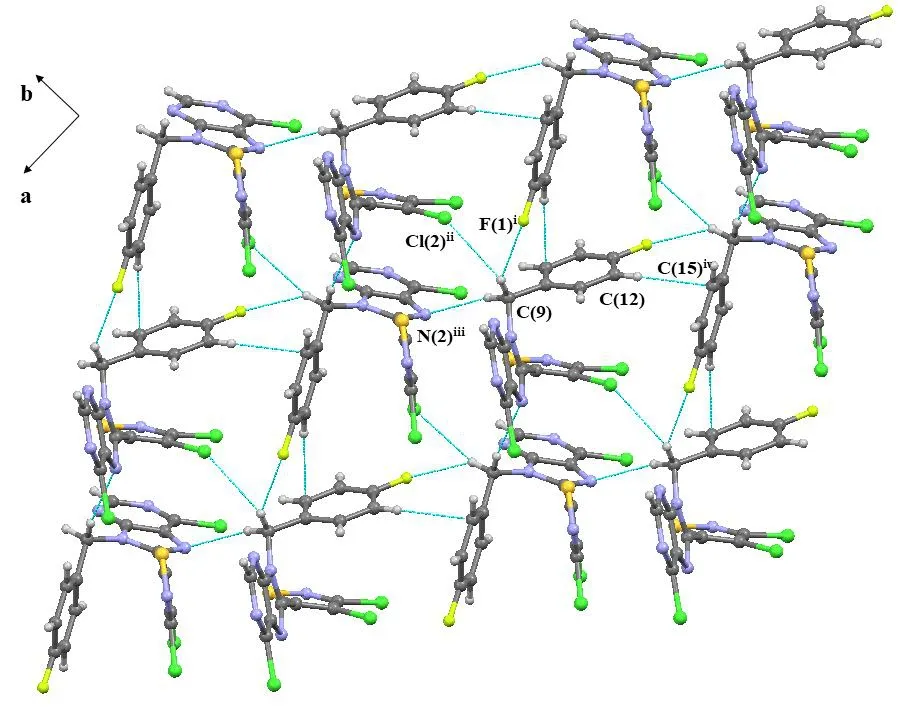
Fig. 3. Crystal packing of compound 3 Symmetry codes: i: -1/2 + x, 1/2 - y, -1/2 + z; ii: -x, 1 - y, -z; iii: -3/2 + x, 1/2 - y, -1/2 + z; iv: 1/2 - x, -1/2 + y, 1/2 -z
Fungicidal bioassay of intermediate 2 and the target compound 3 against seven phytopathogenic fungi at a concentration of 50 mg/L was compared with commercially pyrimidinamine fungicide diflumetorim and lead compound YZK-C22 as positive controls. As shown in Table 4, the intermediate 2 showed weak effects at 50 mg/L, the target compound 3 showed over 50% of inhibitory activities against
B. cinerea,C. arachidicola,G. zeae,R. solani,S.sclerotiorumat 50 mg/L with inhibition of 58%, 53%, 55%,67% and 59%. Most of them were better than diflumetorim but less than YZK-C22. To further assess the fungicidal potency, the EC50values of target compound and positive controls with inhibition over 60% at 50 mg/L were measured.The results shown in Table 5 indicated that compound 3 exhibited good fungicidal activities with EC50value of 25.06 mg/L or 60.44 µmol/L againstR. solani. It was active at the same level of that of the positive control diflumetorim (19.76 mg/L or 60.29 µmol/L) and less active than the positive control YZK-C22 (4.21 mg/L or 12.32 µmol/L)[16]. Docking studies showed that the target compound had larger binding energy with pyruvate kinase than the positive control YZK-C22 because of the effecting of absorption,transduction and metabolism. Our studies indicated that isothiazolopurin derivative could be a fungicidal lead deserving for further study.
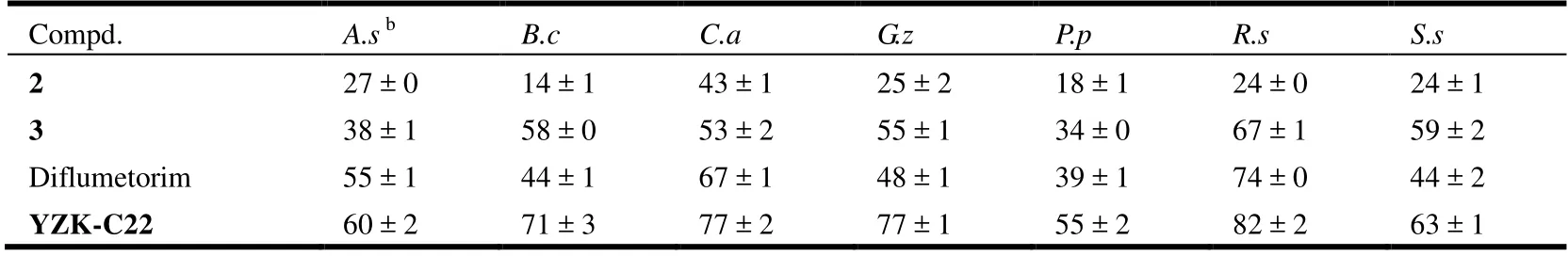
Table 4. Fungicidal Activities of Compounds Synthesized (Inhibition Rate/%)a

Table 5. EC50 of the Target Compounds with Inhibition over 60% at 50 mg/L in Vitro
Dedicated to the 100thAnniversary of Chemistry at Nankai University
杂志排行

结构化学的其它文章
- Synthesis, Crystal Structure and Antifungal Activity of New Furan-1,3,4-oxadiazole Carboxamide Derivatives①
- Syntheses, Crystal Structures, Anticancer Activities and DNA-Binding Properties of the Dibutyltin Complexes Based on Benzoin Aroyl Hydrazone①
- Synthesis, Crystal Structure, Fungicidal Activities and Molecular Docking of Acyl Urea Derivatives Containing 2-Chloronicotine Motif①
- Molecular Design and Property Prediction of High Density 4-Nitro-5-(5-nitro-1,2,4-triazol-3-yl)-2H-1,2,3-triazolate Derivatives as the Potential High Energy Explosives①
- A New Borate-phosphate Compound CsNa2Lu2(BO3)(PO4)2:Crystal Structure and Tb3+ Doped Luminescence①
- Temperature-controlled Structural Diversity of Two Cd(II)Coordination Polymers Based on the Dicarboxylate Ligand①