足细胞能量代谢调节因子与糖尿病肾病
2022-03-09李佳容综述谢红浪审校
李佳容 综述 谢红浪 审校
糖尿病肾病(DN)是糖尿病的常见慢性并发症之一,足细胞功能障碍、脱落和凋亡是DN发生蛋白尿并进展的重要原因[1]。研究发现,足细胞主要通过糖酵解供能,而维持线粒体稳态对足细胞正常发挥生物学功能具有举足轻重的作用[2]。糖尿病时,足细胞胰岛素敏感度下降,线粒体发生氧化应激,自噬功能减退引起足细胞能量代谢障碍和足细胞损伤。AMP激活的蛋白激酶(AMPK)、沉默信息调节因子1(SIRTl)和过氧化物酶体增殖物激活受体γ(PPAR-γ)共激活因子1α(PGC-1α)是足细胞能量代谢的关键调节因子,AMPK、SIRT1级联激活PGC-1α构成AMPK/SIRT1/PGC-1α通路,通过改善足细胞能量代谢,维持线粒体稳态并抑制氧化应激,保护DN足细胞[3]。三者亦可各自通过增强自噬、调节足细胞相关蛋白等作用保护DN足细胞。本文简要介绍足细胞能量代谢调节因子对足细胞的保护作用和DN相关的治疗策略。
足细胞相关能量代谢调节因子
PGC-1α是维持线粒体稳态的关键调节因子,可被多种因子激活,包括SIRT1、AMPK、转录因子EB(TFEB)、丙酮酸激酶M2(PKM2)、牛磺酸上调基因1(Tug1)和肝细胞核因子1α(HNF-1α)等。其中,磷酸化的AMPK和去乙酰化的SIRT1是PGC-1α活化的最强激活物,而参与炎症和纤维化的转录因子抑制PGC-1α的表达[4]。AMPK/SIRT1/PGC-1α通路层级增强PGC-1α的表达并促进PGC-1α磷酸化,改善足细胞能量代谢,并改善线粒体呼吸链功能,增加线粒体密度,从而逆转糖尿病引起的足细胞线粒体功能障碍。
AMPKAMPK是一种丝氨酸/苏氨酸蛋白激酶,是足细胞关键的代谢调节器和能量传感器,由AMP/ATP比值决定其活性,当出现应激、低血糖、缺氧、缺血和运动等ATP消耗增加、AMP浓度升高的情况时,AMPK活化,促进足细胞营养吸收并增强其分解代谢[5]。研究发现,AMPK通过改善足细胞能量代谢,抑制氧化应激,减少足细胞凋亡,促进足细胞肌动蛋白重塑等方式减轻DN足细胞损伤,延缓DN进展。
改善能量代谢 AMPK通过刺激Rac1激活PAK丝氨酸/苏氨酸激酶,以依赖瞬时受体电位阳离子通道(TRPC)的方式激活Rho激酶下游靶,促进细胞骨架重组,进而促进胰岛素介导的足细胞摄取葡萄糖改善足细胞能量代谢[6]。同时,AMPK通过磷酸化乙酰辅酶A羧化酶(ACC)并增加肉碱棕榈酰转移酶1(CPT1)的表达,促进脂肪酸氧化并减少脂质沉积,进而改善足细胞能量代谢[7]。
抑制氧化应激,减少凋亡 AMPK通过下调NOX4-NAD(P)H氧化酶亚基,减少培养小鼠足细胞中超氧阴离子的产生,减少高血糖引起的活性氧产生增加,进而减少足细胞的损伤和凋亡[8]。此外,激活AMPK可抑制高血糖引起的mTOR 信号通路活化,增加足细胞自噬,减少内质网应激,进而减少高糖诱导的足细胞凋亡,延缓DN进展[9]。
促进肌动蛋白重塑 研究表明,AMPK是足细胞肌动蛋白重塑的主要调节因子。闭锁小带蛋白1(ZO-1)是一种紧密连接蛋白,在足突的裂隙横隔膜中高度表达,并将裂隙横膈蛋白与肌动蛋白细胞骨架联系起来,激活AMPK可恢复高血糖引起的足细胞中ZO-1的易位,降低足细胞对白蛋白的渗透性[10]。
SIRT1SIRT1是一种依赖NAD+的脱乙酰酶,作为细胞内营养感受器,通过监测NAD+调节足细胞代谢和氧化还原状态,是细胞衰老的重要调节剂。SIRT1通过维持线粒体稳态,增强足细胞自噬,减少足细胞凋亡,对DN时受损的足细胞发挥保护作用。
改善线粒体功能 高血糖诱导足细胞线粒体功能障碍和凋亡通路激活:糖尿病小鼠肾组织中SIRT1、PGC-1α、核呼吸因子1(NRF1)和线粒体转录因子A(TFAM)的表达随病程延长而逐渐降低,并伴有线粒体肿胀、线粒体嵴紊乱、消失,足细胞形成空泡[11],此时足细胞线粒体ROS生成增加,线粒体呼吸链复合体Ⅰ和Ⅲ活性降低,线粒体膜电位降低,促凋亡因子细胞色素C(CytC)和第二个线粒体衍生的半胱氨酸蛋白酶激活剂/低等电点凋亡抑制因子(IAP)结合蛋白(Smac-DIABLO)释放,含半胱氨酸的天冬氨酸蛋白水解酶3(caspase3)激活,进而促进足细胞凋亡。雷公藤多苷干预后,SIRT1、PGC-1α、NRF1和TFAM表达上调,进而改善糖尿病小鼠足细胞内线粒体形态结构异常及足突融合,并通过改善呼吸链复合物活性,提高线粒体膜电位,抑制线粒体凋亡通路的激活,维持线粒体稳态,进而减少足细胞凋亡[12]。
增强足细胞自噬 自噬是指分解和回收不必要的或功能失调的细胞成分,是一个可调控的过程。足细胞是终末分化细胞,需要高水平的自噬清除细胞内受损的细胞器和活性氧等异常物质。SIRT1是自噬的正调节因子,通过去乙酰化细胞核中的FoxO1和FoxO3以及胞质中自噬体膜形成和伸长所必需的自噬相关蛋白5(ATG5)、ATG7和ATG8来恢复自噬,激活的SIRT1恢复了FoxO3的表达,FoxO3正向调节Bcl2腺病毒E1B19ku相关蛋白3(BNIP3)从而增强db/db小鼠足细胞自噬作用[13]。
抑制氧化应激,减少凋亡 高血糖通过NADPH氧化酶和线粒体途径引起足细胞线粒体ROS合成增多,SIRT1抑制这一过程。同时SIRT1抑制促凋亡因子p38促分裂原活化蛋白激酶(p38 MAPK)和半胱天冬酶3的表达,减少足细胞凋亡[14]。此外,激活SIRT1能够抑制FoxO4的表达,减少 Bcl2L11的表达,减少晚期糖基化终末产物(AGE)异常积聚诱导的足细胞凋亡,进而保护足细胞[15]。
PGC-1αPGC-1α是线粒体生物发生的主要调节因子,多项研究表明,PGC-1α通过促进线粒体生物合成并调节足细胞相关蛋白,减少DN蛋白尿,延缓DN进展。
促进线粒体生物合成 PGC-1α通过上调线粒体DNA增殖和关键线粒体转录因子NRF1/2和TFAM的表达来增加线粒体的生物合成。PGC-1α与PPAR、NRF1/2等转录因子结合促进脂肪酸氧化和调节线粒体动力和能量供应,促进线粒体生物合成,维持线粒体自身稳定[3]。研究发现,PGC-1α靶标过度表达可保护足细胞免受高血糖介导的呼吸复合体活性的降低,线粒体膜电位的改变,维持线粒体稳态,减少糖尿病小鼠的蛋白尿。
调节足细胞相关蛋白 nephrin是裂隙隔膜的组成成分,同时起到信号分子的作用。上调PGC-1α可恢复减弱的nephrin表达进而维持缝隙横隔膜结构的稳定,并促进nephrin与磷脂酰肌醇3-羟基激酶(PI3K)的P85调节亚基结合,刺激足细胞中依赖于PI3K的AKT信号,增加足细胞的存活率[16]。此外,研究还发现PGC-1α可能通过恢复足细胞标志蛋白(podocalyxin)正常表达来维持足细胞结构的完整性,延缓DN进展[16]。
DN时足细胞能量调节因子变化
糖尿病时持续的代谢紊乱抑制PGC-1α的活性并干扰下游的信号级联[17],导致足细胞线粒体功能紊乱,足细胞相关蛋白表达减少,引起足细胞损伤,滤过屏障受损,加重DN蛋白尿[18]。
炎症和纤维化的转录因子上调高血糖通过上调包括Toll样受体4(TLR4)和核因子等炎症相关转录因子来降低PGC-1α表达。随着PGC-1α表达降低,肾小球关键足细胞基因nephrin、足突蛋白(podocin)等表达减少,裂隙隔膜和足细胞的结构稳定性被破坏,肾小球滤过屏障受损,从而导致蛋白尿[19]。
Tug1下调Tug1结合于PGC-1α(PPARGC1A)位点的上游,增强Tug1可以增强PGC-1α的表达,同时Tug1与PGC-1α相互作用进一步增加了PGC-1α的表达[20]。高血糖通过减少Tug1的长链非编码RNA降低了PGC-1α的表达。
PKM2下调实验发现,敲除糖酵解相关因子PKM2的小鼠,其DN症状更为严重,同时,PKM2激活剂导致PGC-1α表达增加,并逆转了高血糖诱导的线粒体功能障碍[21]。即高血糖降低足细胞PKM2的表达,引起PGC-1α表达降低,导致足细胞线粒体功能障碍,引起足细胞损伤。
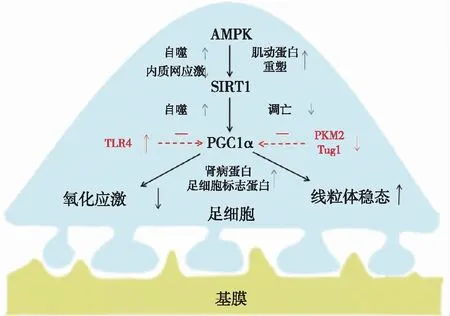
图1 AMPK、SIRT1、PGC-1α保护DN足细胞的机制及糖尿病对其的影响AMPK:AMP激活的蛋白激酶;SIRT1:沉默信息调节因子1;PGC-1α:过氧化物酶体增殖物激活受体共激活因子1α;TLR4:Toll样受体4;NF:核因子;PKM2:M2型丙酮酸激酶;Tug1:牛磺酸上调基因1;红色虚线为糖尿病对足细胞调节因子的改变
治疗策略
二甲双胍二甲双胍是2型糖尿病患者最佳的初始治疗方案,也是各种指南推荐的一线降糖药。研究发现,二甲双胍通过足细胞能量代谢调节因子发挥保护DN足细胞的作用。
减轻氧化应激 NAD(P)H氧化酶是足细胞中负责产生ROS的中心酶,高糖环境下,NAD(P)H氧化酶被过度激活。二甲双胍降低胞外ATP酶的活性,使细胞外ATP浓度增加,激活嘌呤能P2受体,进而激活AMPK并抑制NAD(P)H氧化酶,减少ROS的产生,减轻足细胞氧化应激[22]。
促进足细胞相关蛋白正常表达 其一,二甲双胍使高血糖抑制的nephrin、podocalyxin表达正常化,进而维持足细胞和裂隙隔膜的结构稳定性,并提高足细胞存活率;其二,突触素(synaptophysin)是足细胞中肌动蛋白细胞骨架的基本调节器,二甲双胍恢复了高糖诱导的Synaptophysin减少,减轻了足突消失。其三,DN时钙离子通过TRPC6过度进入足细胞可能导致足细胞损伤和凋亡,在培养的足细胞中,二甲双胍通过激活AMPK使高糖诱导的TRPC6过度表达下调[23]。与此相一致的是,有研究表明在具有Dahl盐敏感背景的链脲佐菌素诱导的糖尿病大鼠中,缺失TRPC6可产生足细胞保护作用。
减轻炎症反应 研究发现二甲双胍降低糖尿病大鼠炎症标志物肿瘤坏死因子(TNF-α)和白细胞介素6(IL-6)的表达水平,减轻足细胞炎症反应,延缓DN进展。
增强自噬保护足细胞 研究表明,二甲双胍通过AMPK/SIRT1-FOXO途径上调SIRT1导致SIRT1底物FoxO1上调,FoxO1易位到细胞核,增强自噬,减少肾脏的氧化应激,减少细胞外基质蛋白的表达并改善肾小球滤过屏障功能[24]。然而,上述研究都没有涉及二甲双胍是否能增强足细胞的自噬,故二甲双胍是否增强足细胞自噬,目前尚不明确。
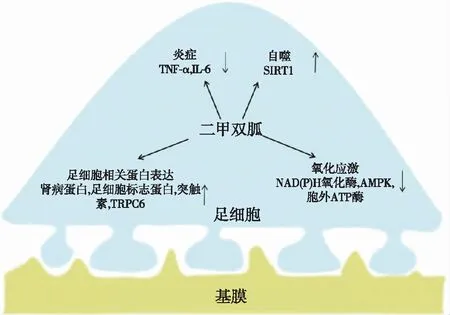
图2 二甲双胍对DN足细胞的保护作用[17]TNF-α:肿瘤坏死因子α;IL-6:白细胞介素6;AMPK:AMP激活的蛋白激酶;SIRT1:沉默信息调节因子1;TRPC6:瞬时受体电位阳离子通道蛋白6
血管紧张素转换酶抑制剂(ACEI)/血管紧张素Ⅱ(AngⅡ)受体拮抗剂(ARB)ACEI/ARB可降低蛋白尿, 改善DN患者预后[25]。研究发现,Ang Ⅱ引起SIRT1 表达失调,并导致SIRT1下游靶标的乙酰化。同时,Ang Ⅱ促进p38 MAPK在DN的足细胞中高表达,加重足细胞凋亡。动物实验表明,奥美沙坦逆转了雄性db/db小鼠SIRT1下调,并抑制p38MAPK在足细胞中的表达,减少了足细胞凋亡[26]。
大黄酸动物实验发现,肾组织中SIRT1的表达与通过高糖高脂饲养联合小剂量链脲佐菌素建立 2 型DN大鼠模型24h尿蛋白定量和肾小球体积呈负相关,大黄酸可通过增加肾组织 SIRT1表达,降低蛋白尿并改善糖尿病大鼠肾脏病理损伤[27]。
雷公藤多苷循证医学研究表明,雷公藤多苷能有效降低蛋白尿、血清肌酐和血尿素氮水平。体内实验研究表明,雷公藤提取物通过维持线粒体稳态,减轻炎症反应进而改善肾小球肥大和足细胞损伤,延缓DN进展,该作用亦在体外实验中得到证实。
改善线粒体功能 雷公藤多苷可上调SIRT1、PGC-1α和TFAM表达,改善线粒体结构和功能,维持线粒体稳态;同时,缺氧诱导因子1(HIF-1)是唯一能在缺氧条件下发挥生物活性的特异性转录因子,参与调节足细胞氧稳态,雷公藤多苷抑制高糖诱导的HIF-1的表达增加,进而减轻线粒体氧耗,抑制足细胞凋亡[28]。
减轻炎症反应 雷公藤多苷抑制TNF-α引起的炎症因子的黏附、聚集和微血管损伤,并抑制依赖NF-κB途径的炎症反应,进而改善高糖诱导的足细胞迁移,降低足细胞蛋白滤过,保护肾小球滤过屏障。
胃肠道反应、肝脏损害、月经紊乱、生殖问题、皮肤不良反应、血液事件、心血管事件和肾毒性等副反应,限制了雷公藤在DN的应用,因此需要优化提取方法,改变其剂型和给药方式,降低毒性[28]。
潜在治疗策略动物研究发现,一些中草药通过改善足细胞能量代谢,改善线粒体功能,抑制足细胞氧化应激等方式,调节足细胞线粒体能量代谢,进而保护DN足细胞。
红景天苷可上调SIRT1、PGC-1α、nephrin和podocin的表达,减轻链脲佐菌素诱导的糖尿病小鼠肾脏结构和功能的损伤,降低了小鼠尿白蛋白、血尿素氮和血清肌酐[29];小檗碱恢复了培养足细胞中CPT1的表达,改善足细胞能量代谢,并通过AMPK/PGC-1α途径增加PGC-1α的表达,改善db/db小鼠和棕榈酸诱导的足细胞中PGC-1α的降低,进而改善足细胞线粒体功能,减轻足细胞损伤[30];此外白藜芦醇改进后的选择性SIRT1激动剂BF175治疗6周,可显著抑制OVE26转基因小鼠(1型糖尿病模型)DN的进展。
小结:AMPK、SIRT1、PGC-1α作为足细胞能量调节因子,构成AMPK/SIRT1/PGC-1α通路通过调节足细胞能量代谢、改善线粒体功能和抑制氧化应激等方式保护DN足细胞。三者亦可各自通过增强自噬、调节足细胞相关蛋白等方式抑制足细胞凋亡。在糖尿病时PGC-1α表达受抑制,二甲双胍、ACEI/ARB、大黄酸和雷公藤多苷等经典药物通过上调SIRT1等因子的表达,维持线粒体稳态,抑制氧化应激来延缓DN进展。一些中药制剂在体外研究中取得结果,但仍需临床研究证实。
