MPI+OpenMP混合并行编程模型在分子动力学中的应用
2022-03-06张坤峰
张坤峰,张 苏
(周口师范学院 计算机科学与技术学院,河南 周口 466001)
近年来,混合编程应用到科学和计算领域,取得了很多成果。何强等[1]针对大规模三维颗粒两相流全尺度模拟,提出了基于MPI+OpenMP混合编程模式。王年华等[2]建立非结构CFD软件MPI+ OpenMP混合并行算法。Wenge Liu等[3]提出了基于OpenMP+AVX的地震并行模拟及优化策略等。以上研究表明,在混合编程模型下,可以加快整个运算的速度,提高性能。
目前在混合编程下研究分子动力学模拟运算的不多,大多数采用MPI进行分子动力学研究。MPI[4]在并行计算MD领域有广泛的应用。LAMMPS大多采用MPI实现并行。本文提出了将MPI+OpenMP混合模型应用到MD模拟中,来提高MD模拟的并行计算性能,设计并实现了LAMMPS软件下基于MPI+OpenMP并行编程模型,实验表明,MPI+OpenMP可以明显提升MD性能。
1 分子动力学模拟分析与软件
1.1 分子运动学模拟分析
分子动力学计算过程如下:
(1)读入指定的运算条件参数,如初始温度、粒子数、密度、时间等。
(2)体系初始化,主要是初始化粒子的位置和初始速度。启动程序,首先要将体系中所有粒子给定初始位置和速度。粒子的位置要相容于模拟体系的结构。粒子不应该放在明显导致计算核心重叠的位置上。选取从一个简单的立方格开始模拟。我们将每个粒子放在晶格上,每个粒子分方向的速度赋予一个随机分布值。接着,改变所有粒子的速度,以确定总动量为零,并标定速度使平均动能为指定值。热平衡时,下式(1)成立,
(1)
其中vα是给定粒子速度α分量。利用这一关系求解在t时刻瞬时温度T(t):
(2)
在计算过程中,不是利用速度来求解牛顿方程,而是根据粒子在当前步的位置和先前步中的位置,以及粒子受到的作用力f,来预测下一时间步的位置。
(3)计算作用于所有粒子上的力。计算每一对粒子i和j在x,y,z方向上的当前距离r。计算粒子间作用时,在距离rc(rc小于半个周期箱)处截断。这样,就可以把i,j之间的相互作用限制在i和j的最近邻映像之间。给定一对粒子足够近以致可以发生作用的话,就要计算这些粒子之间的力和对势能的贡献,假设计算在x方向上的力,那么可以按照公式(3)计算得出,其中r是粒子i跟j之间的距离。
(3)
(4)求解牛顿运动方程。得出下一时刻的位置和速度。 计算完所有粒子之间的力,就要对牛顿运动方程进行积分运算。在程序中,运用Verlet算法,假设在某时刻t,某粒子坐标泰勒展开得,
(4)
同样也可以得到如下算式,
(5)
将(4)和(5)相加,得到,
(6)
根据公式(6)可以得出新位置的坐标。再根据轨迹知识,就可以按照公式(7)导出速度,
(7)
每一时间步结束之后,都要记下新坐标,这样,之前的位置就可以删掉,用最新坐标计算后面的坐标。
(5)整个循环结束之后,计算得出平均值,模拟结束。
1.2 LAMMPS软件
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)[5]即大规模原子分子并行模拟器。LAMMPS是由美国Sandia实验室开发出来的开源软件,用户可以按照自己的需要自行修改代码进行运算。LAMMPS计算MD主要优势是可以在并行环境下运行。
在LAMMPS的运行中,力的计算和创建近邻表占用了大部分时间,本文在设计的模型中主要解决的并行计算部分是Neigh time和Pair time,提高力和近邻表的创建。在每个计算核心上指定一个进程,每个进程将任务分块,然后负责分配到达的任务,每个节点按照自身计算能力,由用户选定线程数,然后派生线程,计算整个模拟过程。
2 MPI+OpenMP并行编程设计
为了满足线程级并行,采用改进安装之后的USER-OMP包中实现函数,保证在运行的过程中能够加入OpenMP,具体方法如下:
(1)设置线程数。通过命令行 export OMP_NUM_THREAD=N; 添加每个进程的线程数。其中,在函数 msm_omp.cpp中会有接收设置的线程数const int tid =omp_get_thread_num(),对于后续采用多线程的运算都是按照变量tid来运算。
(2)构建近邻表的优化。修改neigh_half_
bin_omp.cpp函数,使构建近邻表能够并行处理,在函数void Neighbor::half_bin_newton_tri_omp (NeightList *list)中,加入#pragma omp parallel default(none) shared(list),其中list就是一个共享数组,后期为了避免数据竞争,还要加入critical (list)语句,修改完成之后,加入的N个线程能够并行处理。在构建近邻表的时候,每一个线程开始单独计算粒子近邻表,统一判断是否超过了临界点。如果没有超过,相应的粒子个数就要加1, npage += nthreads。判断完成之后,将得到的近邻表个数传递到相应的粒子列表list > add_pages (nthreads)。
(3)计算力的优化。在pair_lj_cut_omp.h下,定义专门用于计算力的类PairLJCutOMP,在pair_lj_cut_omp.cpp函数中,有专门计算力的类的函数实现方法,将力的计算进行并行处理,在void PairLJCutOMP::compute(int eflag, int vflag)函数之前加入#pragma omp parallel default(none) shared(eflag,vflag),通过get_thr(tid)函数得到要参与计算的线程数,由PairLJCutOMP:compute
(int eflag, int vflag)并行计算每个粒子受到力的情况,通过上面并行语句参与计算粒子在不同方向上各个力的大小。 在力的计算过程中,对于每一个粒子来说,首先inum = list->inum;接收近邻表中粒子的个数,然后对每个粒子都采用牛顿第二定律来计算作用力,其中每个粒子计算三个方向的作用力。
3 测试结果与性能分析
为了说明设计的混合模型满足计算需求,达到计算目的,本文测试环境如下:浪潮服务器十台计算节点(Intel(R) Xeon(R) CPU E5-26200 @ 2.00GHz(12核)和64G内存),一台控制节点,华三1Gbps交换机2台。操作系统为Red Hat Enterprise Linux Server release 6.3,内核版本 2.6.32,MPI环境为openmpi-1.2.6 版,LAMMPS版本是LAMMPS-22Mar13。 对构建的三维坐标30*30*30晶包,原子个数为108 000进行分子动力学模拟,势能选取最简单的Lennard-Jones势能,模拟步数2000步。在LAMMPS下计算MD所用的时间表。
从表1测试的结果可以看到,在作用力计算及构建近邻表两部分中,模拟时间占了整个计算过程的96%。从而说明我们在计算模拟时修改这两方面的必要性。

表1 串行环境下的运行时间(单位:秒)
3.1 混合模型下程序计算时间
为了说明混合模型在LAMMPS下能够很好的运用。首先确定线程数N,测试的节点为12核CPU(原则上每个节点内线程数不能超过12,为方便,此时N=2,2≤N≤12),通过增加节点来确定进程数逐渐增加。首先建立节点间的通信,节点CU01启动mpd,查看节点端口号,之后的所有节点以第一个节点为中心,建成通信环,测试本文设计模型的加速比,结果如表2所示。
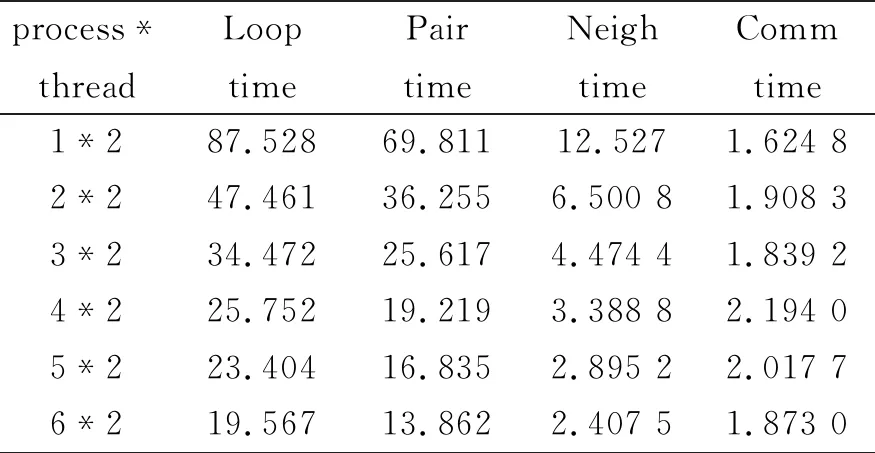
表2 线程为2时的运行的时间(单位:秒)
用加速比计算公式Tp=Tserial/Tparallel来求解表中得出来总的运行时间,在采用MPI+OpenMP混合模型下,LAMMPS在运算中能够达到的加速比如图1所示,

图1 多层并行下的加速比
从表2运行时间来看,当参与计算的节点数为1的时候,会有2个线程在节点CU01下运行,每个线程占用一个CPU,线程之间采用共享内存的并行模式,其加速比可以达到1.94;在多线程确定的情况下,随着进程的不断增加,计算速度在不断加快。当进程数为6的时候,也就是在节点数为6的时候,加速比可以达到8.68,后期因为构建近邻表和力的计算时间所占的比重降低,从而导致整个加速比增长较慢。但是基本上呈现线性增加,这说明本文设计的MPI+OpenMP混合模型在计算MD时具有很好的扩展性。从而表明我们整个模型设计的意义。
为了说明修改近邻表构建和力的计算效率提高,将这两部分的运行加速比作对比。如表3所示,求解上表得出的加速比,采用修改之后的程序运行,其加速比可以用图2表示。

表3 力的计算和近邻表构建加速比

图2 力的计算和构建近邻表加速比
从图2可以看出,修改完成之后,这两部分加速比效率在不同阶段能够最低能够达到80%以上。后期效率有所降低跟选取的计算粒子数量有关系,从加速比的趋势可以得到,本文设计的并行算法适合MD运算。
3.2 单节点下程序运行时间
为了说明我们设计的混合模型具有很好地扩展性。重复以上命令,设置不同的线程数,在单节点下一个进程分配不同线程数进行测试,结果如下表4所示。
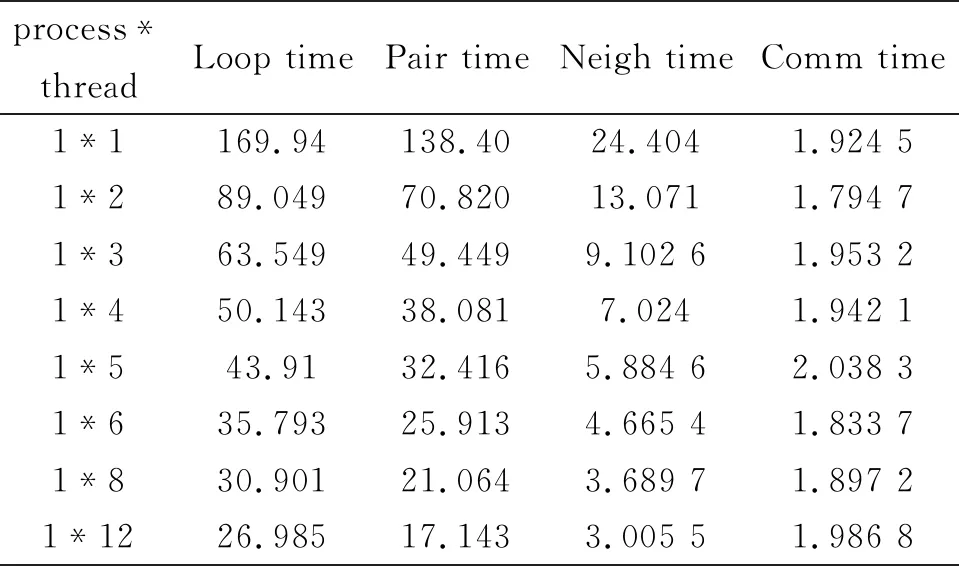
表4 不同线程下运行时间(单位:秒)
利用加速比计算公式,在单节点下,只有OpenMP参与计算,加速比如图2所示。
从图3可以看出,在单节点下,当体系有2个线程,加速比能够达到1.91,分配的2个线程可以并行运行,采用OpenMP实现并行,很好的调度2个CPU参与计算。当分配12个线程之后,加速比达到6.3。这是因为测试的服务器是12核,分配12线程数产生12并行计算线程,从而达到最高值。单进程在内核数的限制下,线程数的增加,使得MD模拟时间减少。测试在单节点下线程数大于12的情况,加速比相对12核来说,非但没有增加。反而停留在线程数为8下的加速比左右,这是因为如果服务器超过线程运算数量,就会采用服务器默认的节点线程数进行运算。进而说明未引入MPI时,在单个节点下,OpenMP的扩展受到服务器核数的制约。
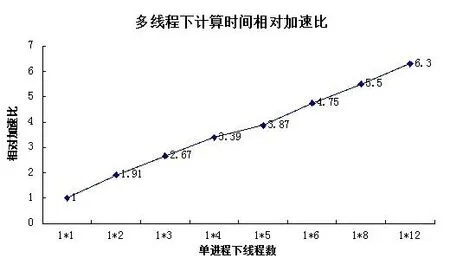
图3 单节点下多线程加速比
3.3 线程数不变的运行时间
为了对比线程的通信时间与进程时间的差别,也就是为了说明节点之间的通信时间的大小与节点内共享内存通信时间的比较。按照在三个节点下,每个节点分配三个线程,以及在九个节点下,每个节点分配一个线程进行测试,测试结果,如表5所示。
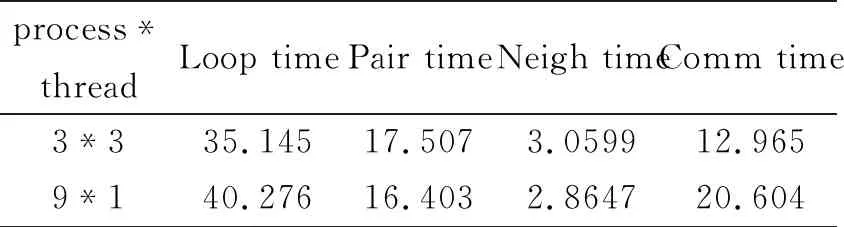
表5 总数不变的运行时间(单位:秒)
根据上表,把每部分计算时间作对比,花费时间如图4所示。

图4 3*3与9*1各个时间对比图
从图4实验结果可以得出,总共有9个线程,总的来看制约分子动力学模拟时间是进程之间的通信时间。首先硬件条件,网络相对于计算机的计算速度以及内存的存储速度来讲,花费的时间会特别多,这也是模拟的一个瓶颈。软件方面,在用OpenMP优化之后通信时间(3*3)所占比例为12.9647/35.1455=36.89%,跟只有MPI(9*1)没有运用OpenMP优化通信时间所占比例20.604/40.276=51.16%小很多。通信时间的增长是因为并发消息条数太多并且每条消息的数据量较大造成的,它们对通信资源的剧烈竞争导致网络拥塞。混合模型虽然增加了单条消息的大小,但是OpenMP优化之后,由于成倍减少了并发消息条数,在单节点下共享内存交换数据,反而减少了节点之间的通信时间,进而说明线程的通信时间比进程通信所花的时间短。从而也满足并行设计中宁可增加计算时间(Pair time),也要减少通信时间(Comm time)原则。在并行设计中尽管在引进多节点混合并行下,OpenMP优化后能够减少进程之间的通信时间,结果表明优化后的效果更能减少模拟时间。
测试完成之后,分析并行模型设计原则,首先采用MPI+OpenMP混合模型,其中加速比能够到达到预期要求,并且其加速比不因为节点计算资源有限,而停止增加,很好的满足扩展性问题。其次,本文采用混合模型,其加大了节点内的并行计算时间,降低通信时间。
综上所述,MPI+OpenMP混合并行编程模型嵌入LAMMPS中,MPI可以增加计算分配线程的数量,使MD模拟计算具有很好的扩展性;在每个MPI进程下,引入OpenMP优化之后,使每个节点之间的通信时间大大降低,缓解MPI节点之间的通信时间过长的不足,这也符合我们并行计算满足加大计算时间和通信时间的比重,从而表明MPI+OpenMP混合并行面层模型有利于MD模拟的计算。
