B36团簇组装一维纳米线的密度泛函研究
2022-03-04刘会霞陈文浩段海明
刘会霞, 陈文浩, 马 洁, 段海明
(新疆大学 物理科学与技术学院, 乌鲁木齐 830046)
1 引 言
硼(B)是元素周期表中碳的近邻,单质硼块体具有多种同素异形体[1]. 包含较少(40以下)原子数目的B团簇倾向于形成平面或者准平面的二维(2D)结构,在这些(准)二维结构中三角形框架为其典型结构特征[2,3]. 然而,随着团簇尺寸的增加,纯二维硼团簇中会出现多边形(四、五、六边形)空位[4-6]. 光电子能谱实验和严格的理论计算均揭示出B36团簇为包含中心六方孔的六边形“碗状”准平面弯曲构型,具有高稳定性和较高的(C6v)对称性[7]. Liu等人通过密度泛函理论(DFT)研究,探讨了六方孔出现在B36团簇中心位置的原因,并且表征了B36团簇在热力学和动力学上也高度稳定的[8]. 王来生教授团队的研究表明,以B36团簇作为结构基元扩展形成包含六边形空位的二维B单层结构也是可行的[7].
自从发现碳纳米管[9]以来,纳米线、纳米棒等一维结构材料因其独特的力学、热学、光学、电子输运等特性而备受瞩目[10-18]. 传统上制备纳米线的实验方法诸多,比如汽液固生长法、氧化物辅助生长法和碳热反应法等[19]. 近年来,团簇组装纳米线方法越来越多地引起研究者的关注,以高稳定团簇作为结构单元组成的一维纳米结构材料具有独特的性能和巨大的潜在应用价值[20,21]. 通常,单质纳米线自身的带隙过小,限制了其在纳米电子器件方面的应用,对此,可以通过掺杂和化学修饰等方式改变原有纳米线的电子结构[22],这对于提升纳米线在纳米器件领域的应用具有明显的重要性和可行性[23,24].
尽管目前关于团簇组装纳米线的研究成果已有诸多报道,但是以B团簇作为结构单元组装纳米线的相关研究还很少见[25]. 据我们调研,至目前尚无B36团簇组装纳米线的相关研究工作发表. 考虑到实验上B36团簇已经成功合成[7]、理论研究也表征了B36团簇具备高的热稳定性[8]和特殊的键合特性[26],本文即采用第一性原理计算方法系统研究了B36团簇组装一维纳米线体系的几何结构与电子结构.
本文中,首先计算分析了孤立B36团簇的电子结构,而后研究了根据不同链接方式组装B36团簇形成的两类不同纳米线,发现二者能量近简并、但分别显示出半金属和小带隙半导体特性,且二者均为热力学稳定体系. 此外,对两种B36团簇组装纳米线进行了H原子吸附,发现吸附后的纳米线均呈现出较大带隙半导体特征.
2 计算方法
采用基于密度泛函理论的第一性原理计算方法. 具体计算使用VASP软件包(Vienna Ab initio simulation package)软件包. 采用投影缀加平面波(PAW)势描述电子-离子相互作用,电子交换关联势选用广义梯度近似(GGA)下的PBE形式[27-30],平面波截断能设置为500 eV,几何结构优化中能量和力的收敛标准分别为10-6eV和0.01 eV/Å. 布里渊区网格密度取1×1×5. 为表征体系的热力学稳定性,采用NVT系综,在室温300 K下,时间步长取为1 fs,模拟总时间长度为3.5 ps,使用第一性原理恒温分子动力学方法计算体系的能量变化行为.
3 结果、分析和讨论
3.1 B36团簇
图1中给出了B36团簇的几何结构示意图及分波态密度图. B36团簇具C6v对称性,自内向外各六边形环上独立的B-B键长为1.662 Å(最内层)、1.670 Å(中间层),1.589 Å与1.673 Å(最外层). B36团簇并非为一完整平面构型,而是一个类似于“碗状”弯曲的准平面结构. 计算所得B36团簇的几何结构与之前Piazza等人的研究结果[7]是一致的. 分析图1(c)可见,B36团簇具有1.07 eV的能隙,且靠近Feimi能的占据态主要由B原子的Pz轨道贡献.

图1 B36 团簇的几何结构示意图((a)为顶视图、(b)为侧视图及分波态密度图(c))Fig. 1 The structure diagrams, top view (a) and side view (b), and the partial density of states (c) of B36 cluster
3.2 B36团簇组装纳米线
将类“碗状”B36团簇作为独立结构单元组成一维纳米线初始构型有两种方式:“碗口”同向型(均朝下、如图2(a)所示)或相邻“碗口”反向型(一上一下、如图2(b)所示),可称为I型纳米线及II型纳米线. 两种类型的初始纳米线经优化后结构均发生了较明显的变化,不再是“碗状”团簇的简单连接,而是呈现出不同“波浪式”的纳米线(如图2(c)、(d)所示).
计算B36团簇组装一维纳米线平均结合能如下:
Eb=(nEB-Enano)/n
(1)
式中,EB为单个B原子的总能量,Enano为纳米线的总能量,n为纳米线所含B原子数. 计算所得I型纳米线和II型纳米线的平均结合能均为5.79 eV,表明这两种类型的纳米线具有相同的能量竞争性.
为了探讨两类不同纳米线的热力学稳定性,采用第一性原理恒温分子动力学方法计算了两类纳米线的能量演化行为. 基于NVT系综,时间步长为1 fs,总模拟时间为3.5 ps,温度取为300 K. 图3给出两类纳米线的温度-时间和能量-时间变化行为,可见,两类纳米线的能量各围绕某一恒定值(~437.5 eV及~438 eV)做小的振荡,均未出现能量的整体(向下)移动. 表明室温下两类纳米线均为热力学稳定的,其动力学演化构型均可以视为相对初始稳定结构的微小偏离.
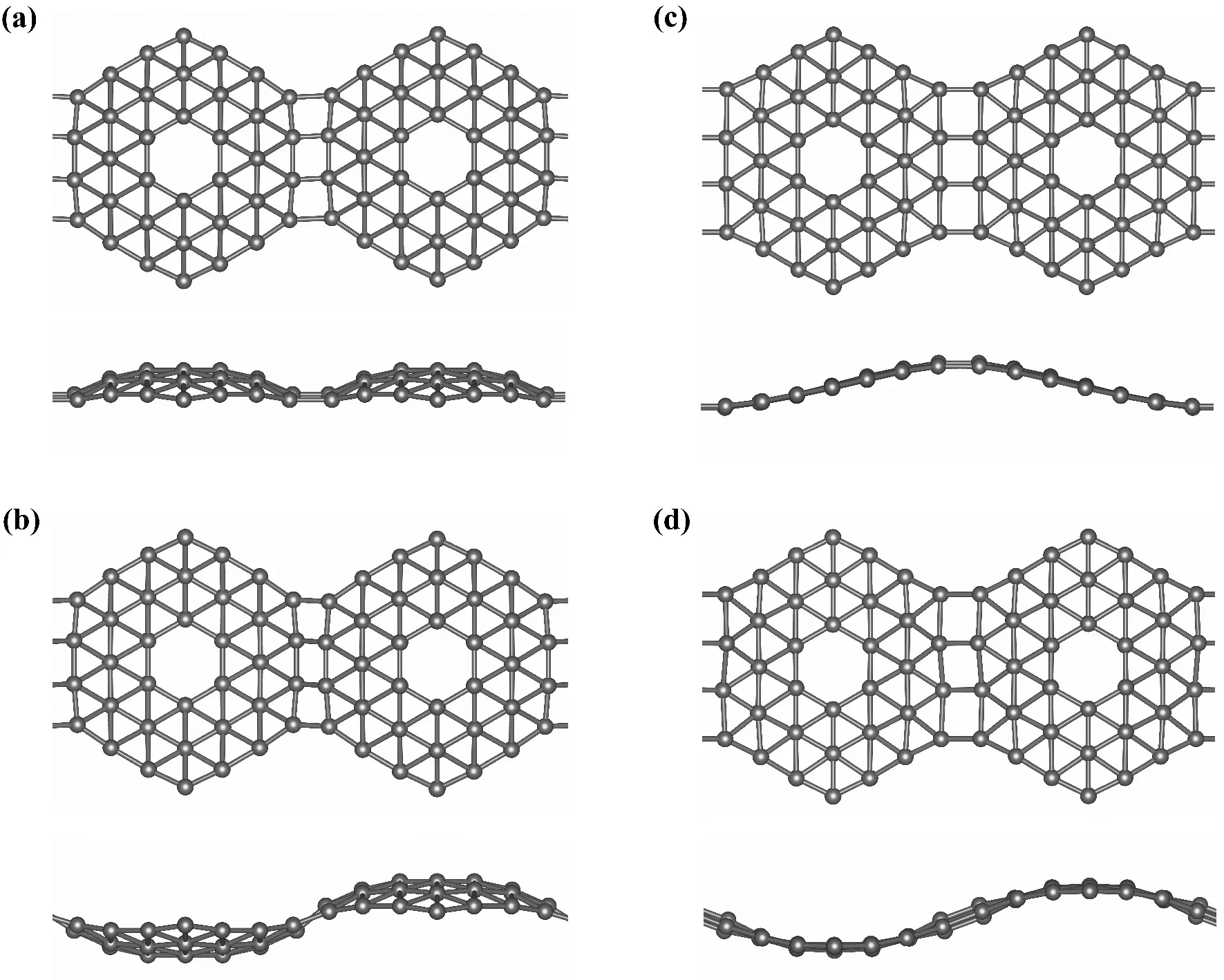
图2 B36 团簇组装一维纳米线的几何结构示意图. (a)、(b)为优化前的初始结构,(c)、(d)为优化后结构. (上为顶视图、下为侧视图)Fig. 2 The structure diagrams of one-dimensional nanowires assembled by B36 clusters. (a) and (b) are the un-optimized initial structures, and (c) and (d) are the optimized final structures. (Top view shown above and side view at the bottom)
为表征B36团簇组装一维纳米线的电子结构,分别计算了两类纳米线的能带结构和分波态密度,结果示于图4中. 由图4(a)可见I型纳米线为一近零带隙的半金属,其费米能级附近电子态主要由B原子p轨道电子贡献,在较深能级(距Feimi能约1eV之外)区域有B原子s轨道电子贡献(见图4(b)). 与I型纳米线明显不同,如图4(c)所示,II型纳米线的能带图呈现出直接带隙半导体特征,具有约0.21 eV的较小带隙. II型纳米线Feimi能附件的电子态密度分布与I型纳米线相类似,如图4(d)所示,费米能级毗邻处电子态主要由B原子p轨道贡献,几乎无B原子s轨道贡献,而在较深能级区间可以看到B原子s轨道的明显贡献. 可见,以两种不同组装方式构造的两类B36团簇组装一维纳米线具有不同的导电性能,分别为近零带隙的半金属和小带隙半导体.
3.3 H原子吸附B36团簇组装纳米线
对于B36团簇组装一维纳米线吸附H原子体系,考虑将H原子吸附在I型及II型纳米线“碗尖”位置B原子上,这与Yu等人研究α-硼烯纳米线吸附H原子的情形是类似的[23]. 图5给出了两种类型的B36团簇组装一维纳米线吸附H原子体系的几何结构示意图,吸附H原子后纳米线的几何结构与未吸附时的结构相较变化甚微,依旧保持原有纳米线的“波浪形”结构,可将其分别命名为I型吸H纳米线和II型吸H纳米线.
以如下公式计算I型及II型吸H纳米线中单个H原子的吸附能:
Eads=(mEH+Enano(B)-Enano(B@H))/m
(2)
其中,EH指为单个H原子的总能量,Enano(B)指为原(未吸附H)纳米线的总能量,Enano(B@H)指吸H纳米线的总能量,m为(计算单元中)H原子数目.
计算所得I型及II型吸H纳米线对H的吸附能均为2.97 eV,体现出二者对H的相同的相互作用.

图3 B36 团簇组装一维纳米线的温度及能量随时间的变化((a)为I型纳米线、(b)为II型纳米线)Fig. 3 Variations of temperature and energy with time of one-dimensional nanowires assembled by B36 clusters for (a) the type-I nanowire and (b) the type-II nanowire
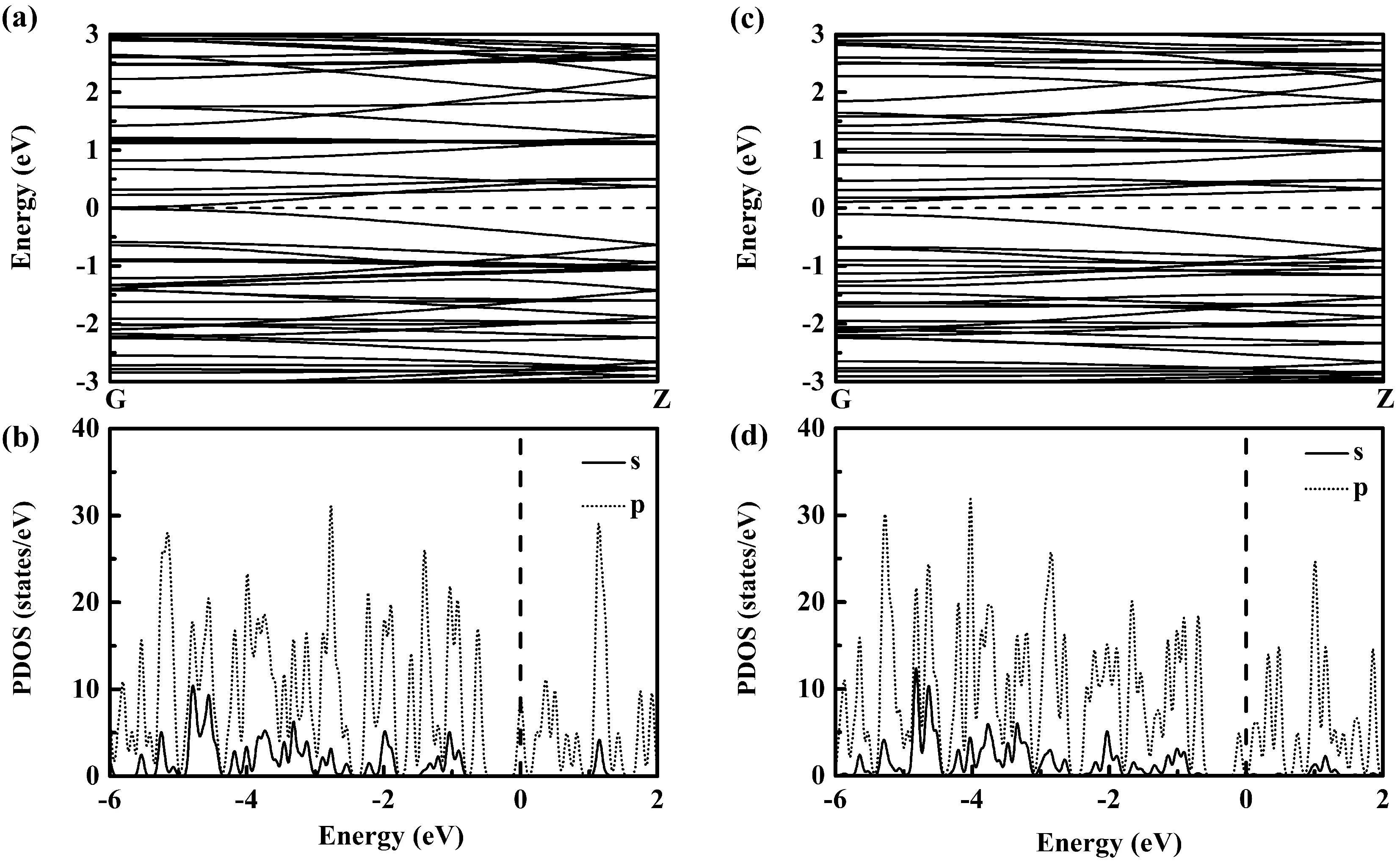
图4 B36团簇组装一维纳米线的能带及分波态密度((a)与(b)为I型纳米线、(c)与(d)为II型纳米线)Fig. 4 Energy band and partial density of states of one-dimensional nanowires assembled by B36 clusters for (a) and (b) the type-I nanowire and (c) and (d) the type-II nanowire

图5 H吸附B36团簇组装一维纳米线的几何结构示意图. (a)、(b)为优化前的初始结构,(c)、(d)为优化后结构. (上为顶视图、下为侧视图)Fig.5 The structure diagrams of H-adsorbed one-dimensional nanowires assembled by B36 clusters. (a) and (b) are the un-optimized initial structures, and (c) and (d) are the optimized final structures. (Top view shown above and side view at the bottom)
与未吸附H纳米线情形类似,为探究吸H纳米线的热力学稳定性,进行了同样(温度300 K、时间步长1 fs、总时长3.5 ps)的分子动力学模拟. 所得I型及II型吸H纳米线的温度及能量演化行为示于图6中.
分析图6可见,在运用恒温分子动力学模拟的初始调温阶段(~500步之前)、伴随着较大的温度涨落两类吸H纳米线的能量变化也出现较大的波动,但在温度趋于恒定后(~500步之后),两类吸H纳米线的能量涨落亦均做微小变化,能量均值基本恒定. 分析该恒温过程中两类纳米线的动力学结构,发现其与纳米线初始结构相比较并无明显变化,这说明两类吸H纳米线在室温下均是热力学稳定的.
H吸附通常会饱和吸附体上相邻原子的悬挂键电子,导致体系电子的重新分布、影响及改变体系的电子结构. 图7中给出了I型及II型吸H纳米线的能带与分波态密度. 原先为半金属的未吸附H的I型纳米线,在吸附H后转变为直接带隙半导体(能隙为0.43 eV,见图7(a)所示). 分析该I型吸H纳米线的分波态密度图(图7(b))可见,邻近Feimi能的占据态(非占据态)主要由B原子p轨道贡献,B及H的s轨道电子贡献很少. B、H的s电子主要分布于较深能级处(-3 eV以下),体现出被吸附H原子与吸附体之间较强的相互作用. 对于未吸附H的II型纳米线,其自身为半导体(具有约0.21 eV的较小带隙),在吸附H后还保持半导体特性,为直接带隙半导体(能隙~0.42 eV,见图7(c)所示). 该II型吸H纳米线的分波态密度结构(图7(d))与I型吸H纳米线相类似:与Feimi能毗邻的占据态及非占据态基本由来源于B原子的p轨道贡献,B、H的s电子主体分布于较深能级处(-3eV以下),也体现出被吸附H原子与吸附体之间较强的相互作用.

图6 H吸附B36 团簇组装一维纳米线的温度及能量随时间的变化((a)为I型吸H纳米线、(b)为II型吸H纳米线)Fig. 6 Variations of temperature and energy with time of H-adsorbed one-dimensional nanowires assembled by B36 clusters for (a) the type-I H-adsorbed nanowire and (b) the type-II H-adsorbed nanowire

图7 H吸附B36 团簇组装一维纳米线的能带及分波态密度((a)与(b)为I型吸H纳米线、(c)与(d)为II型吸H纳米线)Fig. 7 Energy band and partial density of states of H-adsorbed one-dimensional nanowires assembled by B36 clusters for (a) and (b) the type-I H-adsorbed nanowire and (c) and (d) the type-II H-adsorbed nanowire
4 结 论
本文运用基于密度泛函理论的第一性原理计算方法系统研究了B36团簇组装一维纳米线及相应H吸附体系的几何结构、电子结构及稳定性. 以B36团簇最稳定的“碗状”结构为组装单元,按照两种不同对接方式得到两类B36团簇组装一维纳米线. 二者具有相同的能量竞争优势,但二者电子结构显著不同,分别为近零能隙的半金属和小带隙(~0.21 eV)的半导体. 室温下的分子动力学模拟表明二者均为(室温下)热力学稳定体系. 对于两类H原子吸附B36团簇组装一维纳米线,二者均为直接带隙半导体(带隙分别为0.43 eV和0.42 eV),室温下的分子动力学模拟表明二者亦均为热力学稳定体系. 可见,H原子吸附会改变体系的电子结构、增大体系的带隙. 分析吸H纳米线的分波态密度,发现H的s电子基本分布在较深能级处,体现出H原子与B纳米线之间较强的相互作用.
