H2S与CH4重整制氢Mo/Al2O3催化剂的稳定性研究
2022-03-02吴静贤冯昊王昊周君尧张文畅张宏康殷羽彤李平
吴静贤 冯昊 王昊 周君尧 张文畅 张宏康 殷羽彤 李平
华东理工大学化学工程联合国家重点实验室
氢能是一种“零碳”资源与清洁能源,是实现“碳中和”目标的重要载体。预计在未来几十年中,氢能需求将迅猛增长。但是,目前制约氢能发展的一个关键因素是制氢的生产成本[1-3],如何开发出低成本的新型工业制氢技术成为现阶段的一个重要研究课题。统计数据显示,世界上近一半的氢气来源于天然气重整过程。然而,全球范围内约有40%的天然气源中含有H2S,我国的含硫天然气比例更高,占天然气总开采量的70%左右,且大多为硫体积分数超过5%的高含硫天然气,如在四川盆地发现的我国规模最大的普光天然气田,其平均硫体积分数为15%,赵兰庄气田的硫体积分数则高达60%以上[4]。H2S是一种毒性大、腐蚀性强的酸性气体,会对天然气的开采、运输、存储和使用等环节造成严重影响[5],因此,脱除天然气中的H2S是一项长期的艰巨任务。同时,我国进口原油中的硫含量持续上升,经加氢脱硫精制工艺产生的H2S量每年可达数百万吨。因此,处置H2S以符合国家有毒有害污染物排放限制要求所采取的各种措施耗费巨大。
工业上处理H2S最主要的方法是通过Claus工艺将H2S转化为硫磺和水。但相比该工艺将H2S中的氢元素转变成水造成对宝贵氢资源的浪费,H2S直接分解技术则可同步获得H2与硫磺[6-7],因而广受关注。已开发的主要技术路线有直接高温裂解、催化热裂解、电化学分解和光化学分解等,不过这些方法或成本高、能耗大,或装置复杂、效率低,目前仍未有规模化应用的报道[6-10]。
利用H2S与CH4重整反应制取H2是一条全新的制氢反应技术路线[11-12],其反应式见式(Ⅰ)。
2H2S+CH4→4H2+CS2(g)ΔG(1 000 ℃)=-13.68 kJ/mol
(Ⅰ)
2H2S→2H2+S2(g)ΔG(1 000 ℃)=27.61 kJ/mol
(Ⅱ)
与H2S直接分解(式(Ⅱ))相比,H2S/CH4重整制氢反应在相同温度下的平衡转化率高得多,且产氢量大;与CH4水蒸气重整相比,则无CO和CO2排放。该制氢技术路线可利用高含硫天然气或炼油厂加氢脱硫装置产生的大量H2S为原料,能同步获得氢气与高附加值的CS2产品,可在大幅降低H2S与天然气分离成本的同时,充分回收H2S与CH4中的氢、硫与碳资源,符合“原子经济性”原则,并能适应新兴氢能经济发展的需求,因而极具研究价值与应用前景。
迄今为止,国外关于该反应过程的研究报道还不多[11-15]。2008年,Huang等[12]曾对H2S/CH4重整反应过程进行过较全面的热力学分析,认为该反应因需强吸热,须高温才有高转化率,但过程副反应少、能量效率高,且所得产品的商业价值高,所以值得研究开发。同时还指出,唯有降低CH4含量才可避免积炭产生。2015年,Martínez-Salazar等[13]分别以Mo、Zr为活性组分,ZrO2-La2O3和SBA15为载体材料,制备了4种负载型催化剂,应用于H2S/CH4重整反应过程,发现在800~900 ℃、n(H2S)∶n(CH4)=12∶1的反应条件下,反应产物以H2和CS2为主,其中,Mo/La2O3-ZrO2催化剂具有较优的催化活性及抗硫抗积炭性能,其H2S转化率约为1.2%,CH4转化率约为90%,反应在21 h内活性未明显下降,但是反应后的催化剂中检测到了MoS2、ZrS2、MoC及Mo3C2等新物相,而SBA15载体则形成了ZrSiO4混合相,并发生了结构坍塌。Galindo-Hernández等[14]考查了Fe2O3/γ-Al2O3催化剂在950 ℃、n(H2S)∶n(CH4)=12∶1条件下的反应性能,发现在所测试的14 h反应时间段内,H2S的初始转化率高达90%,但4 h后迅速下降至65%,而CH4转化率基本能保持100%,同时,催化剂中的Fe2O3转变成了Fe1-xS。Gupta课题组则报道了H2S/CH4在高温(>950 °C)下发生非催化热解反应的试验结果,发现氢的回收率可达95%,同时,工艺条件对反应转化率影响较大[15]。
在国内,相关的研究尚鲜见报道。本研究对该过程中反应热力学、催化剂制备与硫化条件、反应工艺条件影响等方面进行了考查[16-18],发现当n(H2S)∶n(CH4)=3∶1时,热力学上H2收率最高;以γ-Al2O3为载体制备得到的MoO3/γ-Al2O3催化剂,经硫化后对H2S与CH4重整转化具有很高的活性;但当反应原料中CH4含量较高时,催化剂出现了失活现象。
考虑到含硫天然气中CH4含量一般大于H2S含量,且高CH4含量更有利于H2S充分转化,因此,极有必要研究催化剂在高CH4含量下的稳定性,且目前国内外尚无此方面的研究报道。为此,本研究采用两种不同来源的γ-Al2O3载体,考查所制得的两种Mo/Al2O3催化剂在n(H2S)∶n(CH4)=1∶5、800 ℃条件下的反应稳定性,通过剖析它们在不同阶段的结构变化,揭示催化剂的失活原因,为改善催化剂性能指明方向。
1 实验部分
1.1 催化剂的制备
选取了两种γ-Al2O3载体:商业γ-Al2O3粉末(Com Al2O3),购自国药集团化学试剂有限公司;簇状结构γ-Al2O3(Cluster Al2O3),自行由水热法制备。该载体的制备步骤如下:准确称量九水合硝酸铝(Al(NO3)3·9H2O,AR,上海凌峰化学试剂有限公司)及尿素(CH4N2O,AR,国药集团化学试剂有限公司),加入去离子水搅拌至均相;加入无水乙醇(C2H5OH,AR,上海阿达玛斯试剂有限公司)稀释;转至水热釜180 ℃下加热12 h;冷却至室温后用无水乙醇、水交替洗涤沉淀;随后将沉淀放至真空烘箱中80 ℃下干燥12 h;最后在马弗炉中以2 ℃/min的速率升温至800 ℃煅烧4 h,冷却后得到簇状Al2O3样品。
活性组分的负载采用等体积浸渍法,Mo前驱体为四水合钼酸铵((NH4)6Mo7O24·4H2O,AR,国药集团化学试剂有限公司),浸渍后的样品在500 ℃焙烧4 h,冷却后进行筛分,取40~80目(0.180~0.425 mm)样品进行反应实验。两种催化剂分别标记为Mo/Com Al2O3、Mo/Cluster Al2O3,以MoO3计的质量负载量均为20%。
1.2 催化剂的表征
催化剂粉末样品的晶相结构采用德国Bruker公司的Advance-D8多晶X射线衍射仪(XRD)分析;形貌采用美国FEI公司的Nova Nano SEM 450超高分辨场发射扫描电镜(SEM)观测;微观形貌和尺寸采用日本JOEL公司的JEM2100F的高分辨透射电子显微镜(HRTEM)测量;比表面积和孔径分布采用美国Micromeritics公司的ASAP-2020 低温N2物理吸附仪(BET)测定;热失重采用美国TA公司的TGA5500热重分析仪(TG)分析;积炭量采用德国Elementar公司的VARIO ELⅢ型C/H/N/S元素分析仪分析;拉曼光谱采用日本HORIBA公司的Xplora Plus拉曼光谱仪(Raman)分析;NH3-TPD/CO2-TPD采用美国Micromeritics公司的AutoChem1 II 2920程序升温化学吸脱附仪测定。
1.3 催化剂稳定性测试
催化剂样品的稳定性测试在连续流动固定床反应器中进行,反应器材质为石英管,管内径为8 mm,反应原料气体由反应管上端进入,下端流出,反应尾气经气相色谱仪在线检测后,用乙醇胺多级吸收后放空。采用两台气相色谱仪定量分析反应前后的气体组成,包括H2S、CH4、N2、CS2和H2,其中GC-1色谱仪(Claus 680,美国PerkinElmer公司)以H2为载气、TCD为检测器,色谱柱采用PQ柱(分析H2S和CS2)与5A分子筛柱(分析CH4和N2)串接方式;GC-2色谱仪(Claus 580,美国PerkinElmer公司)采用N2载气、TCD检测器、5A分子筛柱,用来测定H2含量。实验中用到的H2S/N2混合气(体积分数30%)、高纯N2、高纯H2均购自液化空气(昆山)气体科技有限公司,高纯CH4购自上海沃格气体有限公司。
催化剂样品在反应测试前,预先于常压500 ℃及10 mL/min H2S、90 mL/min N2条件下硫化1 h。随后升温到800 ℃,在体积空速为20 000 h-1,10 mL/min H2S、50 mL/min CH4、40 mL/min N2组成的气流氛围中,测定反应出口尾气中的H2S、CH4、CS2及H2含量,以此获得H2S、CH4的转化率,进而计算H2S与CH4的反应速率以及H2的生成速率,两者分别定义为单位质量催化剂上单位时间内转化的H2S(或CH4)和产生H2的物质的量,计算公式见式(1)和式(2)。
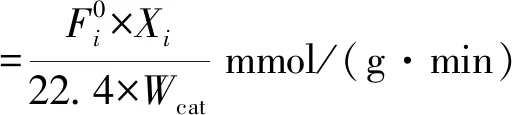
(1)

(2)
此外,对反应前后各物料中的H元素进行了质量平衡计算,结果表明,H元素质量偏差始终保持在5%之内。
2 结果与讨论
2.1 催化剂稳定性比较
图1显示了两种催化剂上H2S和CH4的反应速率及H2生成速率随时间的变化。由图1可以看出,两种催化剂的活性均呈现下降趋势,但程度各不相同。首先,在所测试的20 h反应时间段内,Mo/Cluster Al2O3催化剂上的H2S和CH4反应速率以及H2生成速率都显著高于Mo/Com Al2O3催化剂上的相应结果,特别是H2的初始生成速率(在反应开始后20 min内的结果)达8.4 mmol/(g·min),为后者的3.8倍,相应的H2S初始转化率为76.5%,反映出Mo/Cluster Al2O3催化剂的反应活性优势明显;其次,反应经历20 h后,在Mo/Cluster Al2O3催化剂上,H2S和CH4的反应速率及H2生成速率分别下降了14.2%、17.9%和15.3%,显著低于Mo/Com Al2O3催化剂上各参数的下降幅度,后者分别下降了51.1%、58.3%和53.4%,表明相同反应条件下Mo/Cluster Al2O3催化剂的稳定性明显优于Mo/Com Al2O3催化剂;最后,在两种催化剂上,H2S的初始反应速率与CH4初始反应速率之比,Mo/Cluster Al2O3催化剂为2.5,Mo/Com Al2O3催化剂为2.2,均略高于式(1)中H2S/CH4的化学计量比2,说明H2S与CH4之间发生的反应基本遵循重整反应路径,但也存在着两种反应物各自发生裂解反应的可能性,尤其是H2S裂解生成硫磺和H2。而在反应试验过程中于反应器出口器壁上能观察到少量硫磺附着,证实了部分H2S裂解反应的发生。同时,从两种催化剂上H2S/CH4初始反应速率比值推测,Mo/Cluster Al2O3催化剂比Mo/Com Al2O3催化剂更有助于H2S进行裂解反应。但即便如此,仍然不能排除两种催化剂上CH4发生裂解反应形成积炭的可能性。

2.2 不同阶段催化剂的结构表征
两种催化剂虽然具有相同的活性组分和载体组成,但可能因载体及其负载活性组分颗粒的结构和表面性质存在较大差异,导致催化剂在反应体系中的活性和稳定性表现悬殊。为此,对两种载体和催化剂在不同使用阶段的结构进行了系列表征。
2.2.1SEM表征
图2是SEM照片,揭示了不同载体和催化剂各自的形貌特征,以及催化剂经历硫化与反应后形貌的变化。比较图2(A)和图2(a)可以看出,催化剂Cluster Al2O3与Com Al2O3在形貌上差异显著,前者为许多叶片连接形成的簇状体,结构开放,叶片厚度处于纳米尺度,长度为微米量级[19];而Com Al2O3中的颗粒极不规则,且无序堆积。图2(B)和图2(b)显示了活性组分负载后形成的两种Mo/Al2O3催化剂的形貌。与各自载体相比,催化剂都沿袭了载体原有形貌,没有出现活性组分单独聚集形成的颗粒,意味着活性组分在载体表面高度分散。图2(C)和图2(c)是两种Mo/Al2O3催化剂硫化后的照片。与未硫化样品相比,硫化后的催化剂在形貌上也变化不大,但在总体结构上都有些松散化,出现了少量碎片。当两种催化剂经历20 h反应稳定性测试后,如图2(D)和图2(d)所示,Mo/Cluster Al2O3催化剂的簇状结构存在一定程度的破碎,而Mo/Com Al2O3催化剂的颗粒则出现了团聚结块的现象。

2.2.2XRD表征
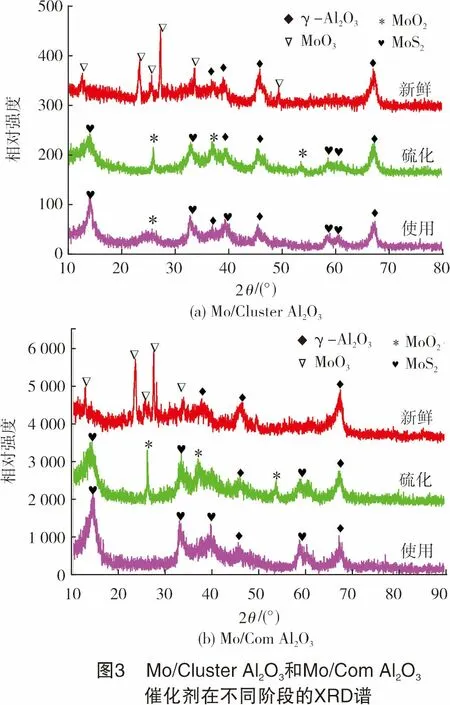
图3是两种催化剂在不同阶段的XRD谱。如图3(a)所示,对于新鲜(未硫化)的Mo/Cluster Al2O3催化剂,位于2θ=37.603°、39.491°、45.862°、67.032°等处的衍射峰与γ-Al2O3标准图谱(JCPDS No.10-0425)一致,其余2θ=12.780°、23.339°、25.699°、27.339°等处的衍射峰则符合层状正交晶系α-MoO3的晶相特征(JCPDS No.35-0609),说明制备得到的新鲜催化剂组成为α-MoO3/γ-Al2O3。比较图3(a)中的3条XRD谱线可以发现,Cluster Al2O3载体在硫化与反应后,仍保持了γ相,显示其良好的耐硫化与耐热稳定性,当然也不能排除有少量高温相如δ相或θ相存在。然而,α-MoO3硫化后转变成了MoO2(JCPDS No.32-0671)与层状六方晶系2H-MoS2(JCPDS No.37-1492)物相,后继连续反应后,MoO2衍射峰减弱,而MoS2的衍射峰增强,说明部分MoO2物种进一步转化为2H-MoS2物种。图3(b)显示,Mo/Com Al2O3催化剂的晶相结构演变规律与Mo/Cluster Al2O3大致相同,只是20 h反应后的Mo/Com Al2O3催化剂中MoO2物相基本消失,同时MoS2的衍射峰更大了。由此可知,反应后两种催化剂的主要组成均为2H-MoS2/γ-Al2O3。由前期研究推测[18],MoO3在硫化过程中可能经历如式(Ⅲ)、式(Ⅳ)的反应,中间相MoO2的出现与MoO3负载量高有关,在反应过程中随着H2S原料气体的持续通入,MoO2会继续被硫化,转化为MoS2物种。此外,由反应后两种催化剂在2θ=58.334°(MoS2(110)面)处的半峰宽计算得到MoS2颗粒的长度[18,20],Mo/Cluster Al2O3约为5 nm,Mo/Com Al2O3约为12 nm,反映出Cluster Al2O3载体上形成的MoS2颗粒分散程度更好。
4MoO3+H2S=4MoO2+SO3+H2O
(Ⅲ)
MoO2+2H2S=MoS2+2H2O
(Ⅳ)
2.2.3TEM表征
两种催化剂上层状结构MoS2颗粒的尺寸大小还可以从HRTEM图上得到信息。图4(A)、图4(B)展示了Mo/Cluster Al2O3催化剂在硫化与反应后表面的微观形态,从中可以清楚分辨出层状结构的MoS2颗粒与Cluster Al2O3叶片基底,其中长条形MoS2颗粒(002)面的层间距为0.65 nm,而Cluster Al2O3基底暴露(110)面上的晶格条纹为(311)面所有,其层间距为0.24 nm。比较图4(A)和图4(B)发现,两者的MoS2颗粒尺寸(长度与厚度)变化不大,约为6 nm×4 nm,说明长时间反应基本不影响MoS2颗粒形态,这可能与MoS2熔点高(>1 800 °C)、热稳定性好,以及载体与MoS2颗粒之间存在强相互作用有关[21]。图4(a)和图4(b)是Mo/Com Al2O3催化剂在硫化与反应后表面的微观图像。由二者可以发现,长时间反应后的MoS2颗粒形态也基本没有变化,长度与厚度的统计平均值为175 nm×5 nm,与Mo/Cluster Al2O3催化剂中的MoS2颗粒相比,尺寸大许多。需要指出的是,由TEM技术观察得到的颗粒尺寸与由XRD技术计算得到的尺寸一般并不相同,前者通常大于后者,这是由两者不同的技术原理决定的,但将两者用于样品颗粒大小比较时,基本可得出一致的结论[18],本研究也是如此。
由于MoS2颗粒边界往往是催化剂活性位所在[22-23],较大的MoS2颗粒上边界活性位占比相对较少,故而推测,Mo/Com Al2O3催化剂活性显著低于Mo/Cluster Al2O3催化剂的原因,在于其活性组分MoS2颗粒明显较大所致。

2.2.4BET和积炭量分析
表1给出了两种载体及催化剂在不同状态下的BET和积炭量测试结果。由表1可知,Cluster Al2O3载体的比表面积明显小于Com Al2O3,负载α-MoO3后对两种载体的比表面积影响较小,样品硫化后比表面积的变化也很小。虽然Mo/Cluster Al2O3催化剂的比表面积并不大,但反应后下降较少,积炭量也很少,而Mo/Com Al2O3催化剂的比表面积在反应后下降了约50%,且积炭量达12%左右。结合上述对两种载体上MoS2颗粒尺寸的分析发现,MoS2颗粒的分散度并不取决于载体的比表面积,而可能与载体的表面化学性质有关。

表1 载体和催化剂的比表面积及反应后催化剂的积炭量样品使用阶段比表面积/(m2·g-1)Vp/(mL·g-1)Dp/nm积炭量,w/%Cluster Al2O3载体70.350.3318.8Mo/Cluster Al2O3新鲜样品65.450.2414.9硫化后70.670.3218.0使用后54.100.2518.50.50Com Al2O3载体106.720.4115.4Mo/Com Al2O3新鲜样品105.530.4215.8硫化后103.830.259.8使用后56.870.1712.111.84
2.3 催化剂失活原因分析
2.3.1TG和Raman分析
为进一步印证催化剂失活主要是由积炭造成的,对反应后的两种催化剂进行了热重分析和Raman光谱测试,得到的结果分别如图5(a)和图5(b)所示。由含氧气氛条件下的TG曲线(图5(a))推断,在200~500 ℃内的失重约为4.6%,是由催化剂中的活性组分MoS2被氧化为MoO3引起的;在500~700 ℃内,催化剂的失重主要是积炭被氧化为CO2所致。很显然,Mo/Cluster Al2O3催化剂上的积炭失重量比Mo/Com Al2O3催化剂上的少得多;在700 ℃之后,两种催化剂的失重基本都是由MoO3物种高温升华造成,由于MoO3负载量较高,因此MoO3升华引起的失重也较明显。

从图5(b)可以看出,位于377 cm-1和403 cm-1两处的拉曼峰是MoS2的E2g1和A1g特征峰,1 580 cm-1则对应碳材料晶体的G特征峰,由碳原子sp2杂化的面内伸缩振动产生,1 330 cm-1处对应碳材料晶体的D特征峰,表示晶体结构中存在缺陷。通常单晶石墨只在1 580 cm-1出峰,而无序碳材料在1 580 cm-1和1 330 cm-1两处都会出峰。Mo/Com Al2O3催化剂上的D峰和G峰十分显著,两者强度之比ID/IG=1.36,说明此类积炭的石墨化程度较低,无定形成分较多[24],而Mo/Cluster Al2O3催化剂上的D峰和G峰几乎观测不到,再次验证了积炭量少的结果。
2.3.2NH3-TPD和CO2-TPD分析
由前所述,载体的表面性质可能对MoS2颗粒的分散及催化剂的稳定性产生重要影响,为此研究了载体的表面酸碱性质。图6(a)、图6(b)分别显示了两种载体的NH3-TPD和CO2-TPD测定曲线,纵坐标单位表示所得结果以样品质量为基准进行了归一化,便于对性能进行定量分析。比较图6(a)中的两条曲线可知,两种载体的NH3脱附峰位置大致相同,在低、中、高温区均有出峰,说明两者的酸强度分布基本一致,都拥有弱、中、强3类酸性位,且以中等强度酸性位居多;Cluster Al2O3的高温脱附峰更偏向高温,反映其强酸性能更强,数量也较多,但在弱、中酸性位数量方面,Cluster Al2O3明显少于同等质量的Com Al2O3载体。从图6(b)图中的两条CO2-TPD曲线看到,Cluster Al2O3在150~350 ℃处的碱性略弱于Com Al2O3载体,而在350 ℃以上,其碱性位数量则较多。
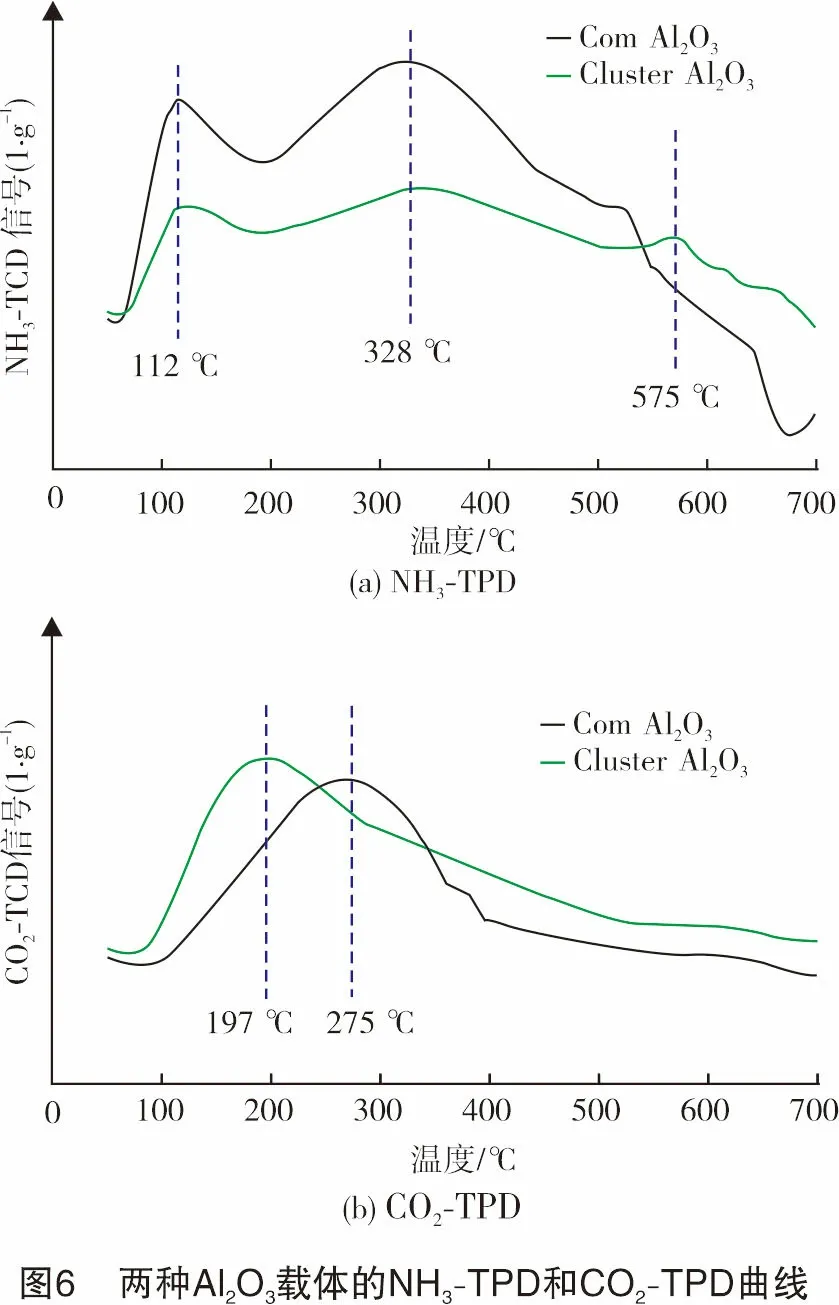
文献研究表明,Al2O3表面的碱性羟基位与酸性位在MoS2前驱体负载过程中起着协同作用[21]。由此推测,虽然Cluster Al2O3载体的比表面积较小,但可能拥有比Com Al2O3载体更适宜的酸碱位强度及数量分布,使得MoS2颗粒在其表面的分散性更好。由于小尺寸的MoS2颗粒边界暴露活性位更多,更有利于H2S快速解离[25],从而形成更多的含S物种加快与CH4之间发生重整反应,因此也降低了CH4单独裂解积炭的风险。此外,载体表面的强酸、强碱位点可能对MoS2颗粒在高温下保持稳定起着关键作用。
综合考虑载体表面性质、MoS2颗粒尺寸与催化剂稳定性,可以认为:载体表面的酸碱性质尤其是强酸、强碱位点数量,对MoS2颗粒的分散与稳定有着直接影响;虽然目前尚不清楚催化剂上H2S与CH4的反应机理,但H2S及CH4裂解反应无疑与重整反应存在着竞争,不同尺寸的MoS2颗粒上反应的竞争程度并不相同,较小尺寸的MoS2颗粒由于边界暴露活性位数量较多,可能更易引发H2S而非CH4裂解反应,而较大尺寸的MoS2颗粒上则可能相反;同时,活性位数量较少的大颗粒MoS2上,积炭覆盖会导致活性位数量减少的幅度更大,因而引起的活性下降就会更显著。
3 结论
对两种不同Al2O3载体制备的Mo/Al2O3催化剂在H2S/CH4重整制氢反应过程中的稳定性进行了比较。20 h反应测试结果表明,Mo/Cluster Al2O3催化剂上,H2S和CH4反应速率及H2生成速率都显著高于Mo/Com Al2O3催化剂上的对应结果,催化剂的活性下降程度也明显较低。对两种载体及催化剂进行的系列表征,揭示了催化剂在形貌、晶相转变、比表面积、活性颗粒尺寸、积炭量等方面的异同。发现与无序化的Mo/Com Al2O3催化剂相比,簇状结构的Cluster Al2O3载体所负载的MoS2颗粒尺寸小,且高温反应后尺寸基本不变,认为与载体表面拥有适宜的酸碱位强度及数量分布,特别是较多强酸、强碱位点对MoS2颗粒起到分散与稳定作用有关;另一方面,小颗粒的MoS2边界暴露活性位多,使得Mo/Cluster Al2O3催化剂不但表现出优异的催化性能,同时其积炭量也显著少于Mo/Com Al2O3催化剂上的量。鉴于反应后两种催化剂上的MoS2颗粒都没有明显长大,因此推断,Mo/Al2O3催化剂活性下降的主要原因是CH4裂解形成的积炭逐渐覆盖活性位所致。
