CYP21A2基因突变致女性假两性畸形一例
2022-03-02朱增哲陈静乐文竹王宏张本平胡蜀红周新荣
朱增哲 陈静 乐文竹 王宏 张本平 胡蜀红 周新荣
患者,42岁,社会性别男,已婚,未育,因“体检发现肾上腺肿物1周余”于2020年11月2日就诊于我院。患者2022年10月22日于当地医院体检,行腹部超声检查发现肾上腺肿块,CT检查提示双侧肾上腺结节灶,无明显不适症状,为求进一步诊治,来我院就诊,以“肾上腺肿物”收入院。患者30年前行尿道下裂手术,父母为近亲结婚,母亲孕期无特殊药物接触史。体格检查:身高150 cm,体重54.5 kg,BMI 24.2 kg/m2,血压120/80 mmHg,身材矮小,男性外观,无喉结,余第二性征(声音、胡须、体毛、阴毛分布等)趋于男性化,乳房无发育,腹部未触及明显包块,无压痛及叩击痛。阴茎短小,阴囊空虚,未触及睾丸(图1)。入院后完善相关检查:血常规、尿常规、大便常规、肝肾功能、电解质、凝血功能、前列腺肿瘤标志物、生长激素、胰岛素样生长因子检查结果均正常。24 h尿钾26.71 mmol/24 h(25~125 mmol/24 h,括号内为正常参考值范围,以下相同);重要内分泌实验室检查结果:17羟孕酮>60.0 nmol/L(3.5±1.2 nmol/L),促卵泡激素<0.20 mIU/ml(1.27~19.26 mIU/ml),促黄体激素<0.20 mIU/ml(1.24~8.62 mIU/ml),孕酮7.83 ng/ml(0.14~2.06 ng/ml),雌二醇27 pg/ml(≤47 pg/ml),泌乳素15.50 ng/ml(2.64~13.13 ng/ml),睾酮3.43 ng/ml(1.75~7.81 ng/ml),游离睾酮指数(FTI/FAI) 65.4%(24.3%~110.2%),游离睾酮0.332 nmol/L(0.110~0.660 nmol/L);雄烯二酮>35.0 nmol/L(3.7±0.9 nmol/L),脱氢表雄酮8 330 nmol/L(5 723±2 380 nmol/L),促肾上腺皮质激素200.2(7.2~63.3 pg/ml),皮质醇(8AM)47.3 μg/L(60.2~184 μg/L),活性肾素浓度28.2 μIU/ml(立位4.4~46.1 μIU/ml,卧位2.8~39.9 μIU/ml),醛固酮184 pg/ml(立位0~353 pg/ml,卧位0~236 pg/ml)。促性腺激素释放激素(GnRH)兴奋试验及人绒毛膜促性腺激素(HCG)刺激试验结果均为阴性。影像学检查结果:睾丸彩超结果提示1.双侧阴囊小;2.阴囊内未探及睾丸回声。肾上腺MRI结果提示双侧肾上腺区结节,右侧大小约28 mm×23 mm,左侧大小约17 mm×12 mm(图2)。盆腔MRI结果提示膀胱直肠间可见子宫,未见明确精囊腺信号。双侧阴囊脂肪填充,未见明显睾丸信号(图3)。染色体核型分析:46XX,del(15)(p11)。基因检测突变位点(图4):CYP21A2,6p21.33,NM 000500.7:Intron2:c.293-13C>G:p.及Extron4:c.518T>A:p.(lle173Asn)。综合患者病史、体格检查及辅助检查结果,诊断为性发育异常:46XX女性假两性畸形,21羟化酶缺乏。我们对患者家族史进行详细询问后发现其存在家系遗传,两个兄弟出生后不久夭折,姨妈、舅舅也患有该病,符合常染色体隐形遗传(图5)。患者目前予地塞米松治疗,补充生理所需皮质醇,生活上无障碍,电解质检查结果正常。患者虽然存在子宫,但未见明确卵巢、输卵管结构,子宫较小,并非正常成人子宫形态,不具备生殖能力,且患者适应“男性”性别42年,故目前依旧保持社会男性性别,未行矫形手术。
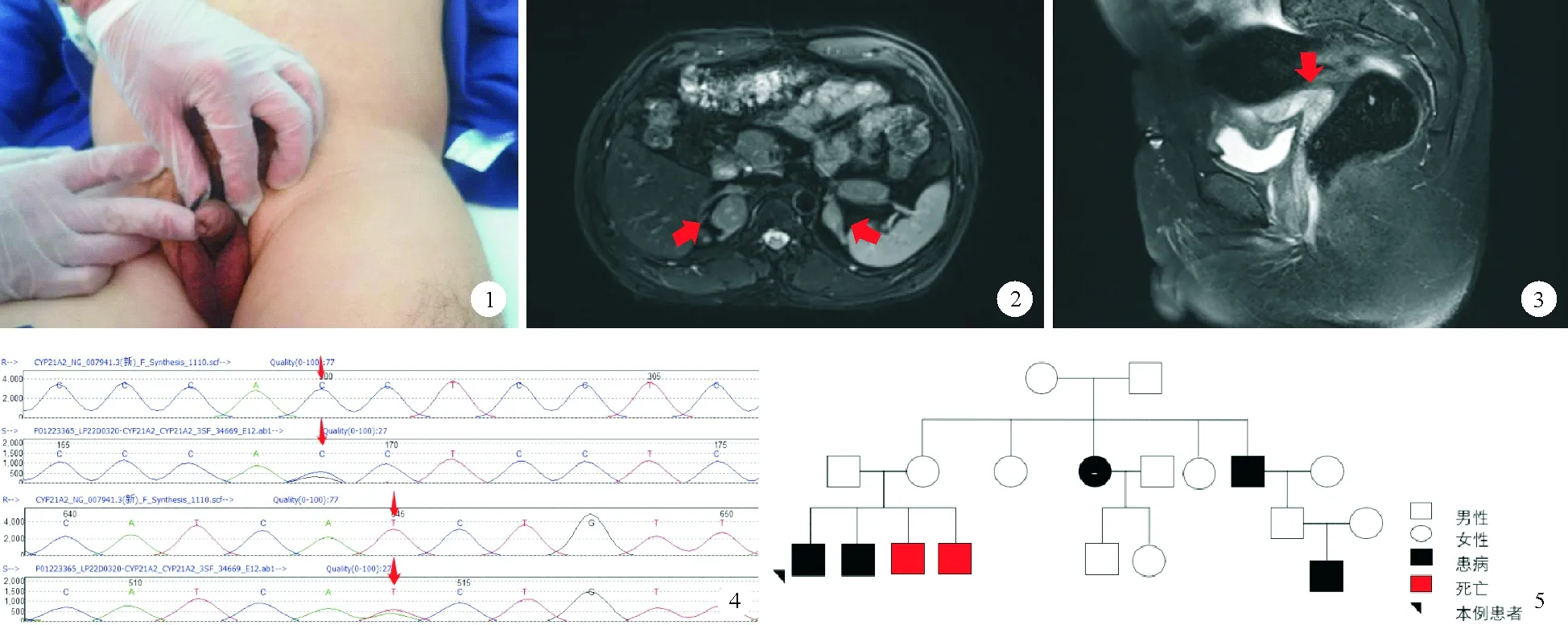
图1 患者外生殖器表现为男性,阴茎短小,阴囊小,未触及睾丸 图2 肾上腺MRI结检查果:红色箭头示双侧肾上腺结节,右侧大小约28 mm×23 mm,左侧大小约17 mm×12 mm 图3 盆腔MRI检查结果:矢状位图像可见膀胱直肠间存在“7”字型子宫形态(红色箭头所示) 图4 患者基因检测结果:红色箭头示基因突变位点 图5 患者家系图
讨 论
先天性肾上腺增生(CAH)是一组由编码皮质激素合成途径的必需酶基因突变,致肾上腺皮质类固醇激素合成障碍所引起的疾病,呈常染色体隐性遗传,是引起两性畸形最常见的病因,其中21羟化酶缺乏(21OHD)最多见,约占所有CAH的90%[1]。
人的21羟化酶由6号染色体p21.3的CYP21A2基因编码,据人类基因突变数据库统计,全球已报道了近300种不同的突变[11]。研究表明,CYP21A2基因的intron2末端前第十三个碱基点突变(c.293-13A/C>G)是中国人最常见的突变,约占所有21OHD患者的42.5%[9],其次是Extron4中的错义突变(c.518T>A,lle173Asn),在中国21OHD患者中的发生频率约为12.7%~20.0%[9-10],而在单纯男性化型患者中,发生频率高至29.1%。c.293-13A/C>G突变可导致RNA剪接异常,使所编码的蛋白质发生紊乱而丧失正常功能[8];c.518T>A,lle173Asn使得所编码的蛋白质第173位氨基酸由异亮氨酸(Ile)变为天冬酰胺(Asn),导致21羟化酶活降低,残留约1%~2%酶活性[6]。本例患者基因突变为Intron2:c.293-13C>G:p及Extron4:c.518T>A:p,是人群中最常见的两种突变类型。
21羟化酶负责将17-羟孕酮(17OHP)转化为11-脱氧皮质醇,将孕酮转化为脱氧皮质酮。由于21-羟化酶功能障碍,皮质醇生物合成途径被阻断,导致前体的积累,其中最显著的是17OHP,因此17OHP也被用于21OHD的诊断[2]。这些前体可以通过旁路途径转化为雄激素。
CAH根据临床严重程度分为3种类型:(1)失盐型:因其21羟化酶活性完全缺乏[6],除了有男性化表现,还有醛固酮缺乏的特征[2]。(2)单纯男性化型:单纯男性化型患者残留1%~2%的酶活性[6],可产生足够的醛固酮,男性可出现进行性阴茎增大和睾丸小,女性出现阴蒂增大,多毛症,男性型秃发,月经异常和不育,表现为两性畸形[3]。(3)非经典型:保留20%~60%的酶活性[7],可以产生足够的醛固酮,只有轻度雄激素过多的临床表现,女性患者在出生时外生殖器正常或轻度阴蒂肥大,没有外生殖器两性畸形[2]。
实验室检查可发现17OHP、促肾上腺皮质激素(ACTH)、血浆肾素活性(PRA)、雄激素、尿17酮类固醇水平升高,皮质醇降低、醛固酮降低。其中17OHP对21OHD诊断有重要意义,当17OHP>30 nmol/L提示21羟化酶缺乏[7,11]。影像学检查有助于判断内生殖器官与外生殖器官的一致性,女性假两性畸形患者一般存在女性内生殖器官,影像学检查可发现子宫、卵巢、输卵管、阴道等女性内生殖器官结构,而不存在睾丸、精索、精囊、输精管等男性生殖器官。另外,CT、MR检查发现肾上腺增生对疾病诊断也有重要意义,不少患者是由于发现肾上腺增生后确诊为21OHD。21OHD患者有男性化表现,染色体核型鉴定可明确其染色体性别,对疾病诊断有重要意义[4,11]。
21OHD的治疗涉及儿科、内分泌、泌尿外科、心理科等多学科合作,包括药物治疗和手术治疗两方面,糖皮质激素是21OHD治疗的主要手段[4]。确诊的患者应尽早行手术矫正治疗,应遵从染色体检查结果及患者、家属意愿,尽早帮助患者行性别选择,减轻心理社会压力。
本例患者因CYP21A2基因突变,导致21羟化酶缺乏,但仍有残留21羟化酶活性,故表现为单纯男性化型。该患者患病与其父母近亲结婚有关,其家系存在明显遗传。因此,早期识别21OHD导致的女性假两性畸形尤为重要,尽早纠正性别畸形,提高患者生活质量。
