氯吡格雷注射剂的研究进展
2022-02-28王雪怡孙平平孙春萌涂家生
王雪怡,孙平平,孙春萌,涂家生
(中国药科大学药学院药剂系,国家药品监督管理局药物制剂及辅料研究与评价重点实验室,中国药科大学药用辅料及仿创药物研发评价中心,江苏 南京 210009)
氯吡格雷(Clopidogrel,化合物1,见图 1)是一种噻吩并吡啶类前体药物,本身无活性。口服经肠道吸收后,85%的药物被肝脏脂酶快速代谢水解成羧酸衍生物(化合物4,见图1),其余的15%经过两步细胞色素P450(主要是CYP3A4、CYP2C19)两步氧化反应,生成含有巯基的活性代谢物(化合物3,见图1)发挥抗血小板作用。含巯基的活性代谢物可特异性、不可逆地和血小板 P2Y12受体结合,从而阻断二磷酸腺苷(adenosine diphosphate,ADP)和受体的结合,阻断ADP诱导的血小板聚集,抑制腺苷酸环化酶,使环磷酸腺苷(cyclic adenosine monophosphate,cAMP)浓度下降,发挥抗血小板作用[1-2]。作为经典的拮抗P2Y12受体的抗血小板药,它和阿司匹林的双联抗血小板治疗已成为防治心脑血管疾病的药物基石,被广泛用于预防急性冠脉综合征(acute coronary syndrome,ACS)患者缺血性事件的复发及经皮冠状动脉介入治疗(percutaneous coronary intervention,PCI)后支架内血栓的形成。
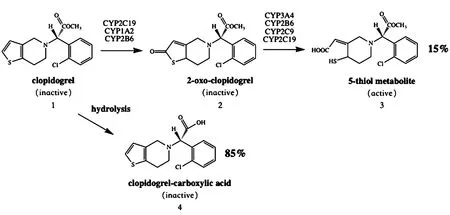
图1 氯吡格雷的代谢途径
硫酸氢氯吡格雷片(商品名为Plavix,波立维)是Sanofi和Bristol-Myers Squibb联合开发的第二代P2Y12受体抑制剂类抗血小板药物,1998年6月首次在美国批准上市,2001年在中国批准上市,规格包括每片75 mg和300 mg。氯吡格雷的常规剂量是每天75 mg,对于ACS患者可采用负荷剂量的方法,即首剂口服300 mg,此后每天75 mg维持。单剂量口服氯吡格雷75 mg后,氯吡格雷的半衰期为6 h,活性代谢产物的半衰期约为30 min,患者口服 3~7 d内可达到稳态血药浓度[5]。
近年来,中国开展PCI的数量逐年上升,2019年已经超过100万例[6],但对ACS患者的急诊处置水平和发达国家仍有不小的差距。在PCI手术前给予负荷剂量的氯吡格雷,可显著减少心血管事件发生率和明显改善患者PCI术后的主要心血管预后,改善手术后缺血。而目前临床上可以使用的氯吡格雷剂型仅有片剂,其口服后起效速度慢,对急需手术治疗的ACS患者常面临错失最佳治疗时机的风险;此外,患者需要口服较大剂量氯吡格雷才能达到血液中有效药物水平,在提高抗血小板速度的同时可能带来出血等风险[10-11]。而解决以上问题的关键在于如何建立快速预防支架内血栓形成和剂量过高造成出血风险之间的平衡[7,12]。与口服剂型相比,直接静脉注射抗血小板药物不仅可以实现临床治疗时的快速起效,还可以有效缩短停药后的药物作用时间,能够在兼顾有效性和安全性的前提下为医生提供更多的临床用药选择。
2015年3月和6月,EMA和FDA分别批准了一种P2Y12受体可逆性抑制剂抗血小板注射剂坎格瑞洛 (Cangrelor) 上市。坎格瑞洛是首个静脉使用的抗血小板药物,其具有起效迅速、失效快、作用可逆等特点,具有一定的临床优势。然而,由于可能引发临床出血并发症,坎格瑞洛仅被限制作为二线药物用于之前未接受口服 P2Y12抑制剂患者的PCI手术和口服治疗不便的患者[15],其治疗ACS的效果还需要更多的临床研究支持。而氯吡格雷作为目前应用最广泛的P2Y12受体抑制剂,其注射剂的开发和应用极具临床意义。
近年来,包括氯吡格雷口服制剂的原研公司Sanofi在内,有多家公司对氯吡格雷及其盐进行注射剂型的开发 (见表1)。本文将综述国内外氯吡格雷注射剂的研究情况,介绍其产品特点、制剂技术以及临床应用等,并对此类制剂的药学研究内容进行分析。

表1 氯吡格雷及其盐进行注射剂型的开发
1 氯吡格雷注射剂型的研制
氯吡格雷游离碱及其硫酸氢盐的溶解性具有高度的pH依赖性,其在生理条件(pH 7.4)下的溶解度仅为0.002 mg·mL-1,随着pH降低溶解度不断提升,当pH为1时溶解度可达7 mg·mL-1[16],但该pH已不适合于直接注射;此外,氯吡格雷为前药,给药后需在体内被代谢成化合物3(见图1)才具有抗血小板活性。因此,氯吡格雷注射剂型的开发重点和难点均聚焦于解决以上两个问题。
1.1 氯吡格雷冻干粉针 氯吡格雷及其片剂的原研公司法国Sanofi曾投入研发注射用氯吡格雷粉针剂,以弥补口服氯吡格雷片在临床急救上的不足。氯吡格雷及其硫酸氢盐单独冻干时,容易形成不溶性的聚集物,黏于玻璃壁上。根据Sanofi公司的专利,在制剂中加入泊洛沙姆188可阻止氯吡格雷及其硫酸氢盐的聚集[17]。将氯吡格雷及其硫酸氢盐和泊洛沙姆188的水溶液冻干后,用特定溶剂(由聚乙二醇硬脂酸酯Solutol HS15和磷酸缓冲盐调配而成,pH调至4.0以上)复溶后再注射。专利中还显示,在冻干粉中采用甘露醇和丙氨酸可提高药物稳定性[18]。但最终,Sanofi研发团队并没有继续该项目的开发,原因未知。
1.2 氯吡格雷β-环糊精包合物注射剂(MDCO-157) MDCO-157由Ligand Pharmaceuticals公司的子公司CyDex Pharmaceuticals开发。早期该项目由Prism Pharmaceuticals公司开发用于治疗心血管疾病。2011年,Ligand从Prism Pharmaceuticals公司获得开发许可,并更名为MDCO-157[19]。同年Medicines公司从Ligand公司获得许可开发用于抗动脉粥样硬化血栓形成,在美国开始抗血栓治疗的I期临床试验。
MDCO-157采用了CyDex Pharmaceuticals Inc研发的Captisol磺基烷基醚环糊精(SAE-CD)与氯吡格雷游离碱形成包合复合物。药物与SAE-CD 的可逆、非共价复合可提高其在水溶液中的溶解度和稳定性。利用SAE-CD可以有效改善氯吡格雷溶解性差的问题,将氯吡格雷增溶至20 mg·mL-1,并且减少溶液中氯吡格雷的化学降解和手性转变[20]。
MDCO-157的开发旨在其可以与口服氯吡格雷产生相当的抗血小板作用,从而可以从口服过渡到静脉给药来改善高危PCI患者的临床治疗。然而,一项2012年开展的临床试验(33位受试者,氯吡格雷口服制剂作为对照,随机开放标签交叉试验)结果显示,虽然健康志愿者的安全性和耐受性良好,但由于活性代谢物(见图1中化合物3)产生阈值不足,当静脉给药剂量增加到300 mg时,也不能达到足够的血小板抑制作用。MDCO-157在静脉注射75、150和300 mg剂量组中表现出剂量相关的药效学效应,但与口服氯吡格雷300 mg剂量相比,静脉注射MDCO-157显示短暂而轻微的药效学效应,巯基活性代谢物的Cmax和AUC低于口服氯吡格雷300 mg剂量。而且注射剂的不良反应非常大,包括输液疼痛、头痛、头晕、血管穿刺部位血肿等[19]。
1.3 氯吡格雷纳米乳注射剂(ASD-002) ASD-002是Ascendia Pharmaceuticals开发的一种氯吡格雷纳米乳注射剂型。2016年11月,Ascendia公司和FDA就氯吡格雷纳米乳注射剂的临床申报工作举行了Pre-IND会议。2020年8月,乐明药业(苏州)宣布与Ascendia Pharmaceuticals签订合作协议,获得氯吡格雷纳米乳注射剂(ASD-002)的开发许可。
ASD-002作为Aascendia公司的主要管线产品是利用其EmulSolTM纳米乳技术平台开发的,实现了经典抗血栓药物氯吡格雷的可注射形式。利用Ascendia的EmulSolTM技术可生产稳定的纳米乳,而无需使用有机溶剂。EmulSolTM采用了传统的高压均质工艺,通过选择特定的长链甘油三酯和离子型表面活性剂最大限度地减少了助表面活性剂的使用。纳米乳平均液滴大小约为200 nm,使用大豆油和表面活性剂卵磷脂的专利组合[16],由此产生的水相中的油滴悬浮液在物理上稳定,并且提高了给药的安全性。
纳米乳剂能够使可溶性差的药物用于多种给药途径,并且能够潜在地保护活性成分免受化学降解[21]。氯吡格雷游离碱为高黏性半固体的油状物,具有化学不稳定、易水解和氧化、手性中心的质子不稳定等问题[16]。由于手性中心和甲酯基团中存在不稳定的质子,因此它非常容易发生甲酯基团的外消旋、氧化和水解。根据专利显示,纳米乳可以有效抑制氯吡格雷从S对映体(有生物活性)到R对映体(无任何抗血小板聚集活性)的转化[16]。该纳米乳技术将氯吡格雷游离碱分散在油相中制成油/水型乳剂,即使游离碱在血浆pH条件下亦很难溶解,当包含在纳米乳液的油相中,载药量可达到20 mg·mL-1以上,改善了难溶性药物的溶解度低的问题[16]。
1.4 氯吡格雷纳米脂质体混悬剂(JIN-2013) JIN-2013是由Intas和Jina制药公司联合开发的静脉注射氯吡格雷纳米脂质体混悬剂,活性成分为硫酸氢氯吡格雷,并采用了Jina公司专有的NanoAqualip脂质纳米技术进行制备,该方案能够在不使用有机溶剂静脉注射的情况下制备出具有良好性状的氯吡格雷脂质体制剂[22-23]。据文献报道,氯吡格雷纳米脂质体混悬液采用全自动高压均质机进行制备,粒径在25~110 nm,辅料包括:大豆磷脂酰胆碱、胆甾醇硫酸钠、蔗糖、缓冲剂[24]。2015年10月,该项目已完成Ⅰ期临床研究(Lambda Therapeutic Research Inc)。
临床前相关药代动力学研究结果显示,注射氯吡格雷纳米脂质体混悬液的Tmax和Cmax(0.5 h和38.0 μg·mL-1),与口服硫酸氯吡格雷(2 h和20.4 μg·mL-1)相比具有更短的Tmax和更高的Cmax[24],证实了其在临床急性给药方面的优势。在一项48例的健康受试者参与的Ⅰ期临床安全性研究中,研究者比较了氯吡格雷片剂300 mg、氯吡格雷纳米脂质体混悬液注射剂25、50和75 mg剂量的安全性,结果表明剂量至75 mg仍安全和耐受[24]。在临床研究中发现,该制剂对口服氯吡格雷无药效的受试者可产生治疗效果[24],与氯吡格雷传统剂型相比,体现了纳米脂质体混悬剂作为药物递送系统在肠外给药方面具有的一定优势。
2 氯吡格雷注射剂开发的探讨
近年来,抗血小板类药物注射剂的开发已经成为行业的研究热点,而氯吡格雷在临床上迫切需要被开发成一种能快速起效的剂型,注射剂成为必然首选。然而由于氯吡格雷自身理化性质及其在体内作用特点的特殊性,大大提高了将其开发成注射剂型的技术难度[16]。
首先,氯吡格雷在中性pH条件下溶解性极差,使其与体液接触时易产生沉淀,从而导致注射痛、静脉炎,甚至可能在给药过程中造成栓塞。其次,氯吡格雷游离碱在潮湿和高温的情况下不稳定。由于氯吡格雷是一种手性分子,其可以作为R或S对映体存在。S对映体具有生物活性,而R对映体无任何抗聚集活性并且耐受性差[25],在动物体内高剂量引起惊厥。因手性中心存在不稳定的质子,氯吡格雷游离碱结构并不稳定,容易发生外消旋、氧化和甲酯基水解[26]。化学不稳定性限制了氯吡格雷在处方中水溶液的使用,使其处方条件局限为含有机溶剂的液体或冷冻干燥固体,其储存条件限制于低温冷藏或冷冻。
对于难溶药物注射剂的研制,科研工作者已经给出了多种解决方案[27]。目前,制备处方中含有难溶性碱性药物的静注或口服液体制剂有多种方法,包括纳米混悬剂、通过环糊精及其衍生物制备包合物、纳米乳、以及在溶液低pH情况下与强酸形成盐等[28],其中多数已在氯吡格雷注射剂的开发中予以尝试。然而,对于纳米混悬液系统,由于其中纳米级药物颗粒在水中的暴露面积较大,可能会加速主药的降解[28]。此外,由于水溶液中的游离药物浓度较高,注射疼痛可能是纳米混悬系统的另一个问题。对于纳米乳,其达到稳定需要高浓度的表面活性剂和助表面活性剂并且稳定性受温度和pH影响[29]。环糊精及其衍生物可能引起潜在的肾毒性、心动过缓和血压降低,以及环糊精可能与合用亲脂性药物结合的问题[30]。而硫酸氢氯吡格雷等强酸形成的弱碱性盐溶液pH值较低,可能导致药物稳定性问题。当在中性pH条件下与血液接触时,药物可能沉淀为游离碱导致注射部位刺激和疼痛。
近年来,涂家生等[31]设计开发了采用一种以mPEG-PLA为载体的氯吡格雷胶束。以两亲性嵌段共聚物mPEG-PLA和氯吡格雷游离碱通过薄膜分散法制备得到胶束纳米系统,发挥了胶束高溶解度、快速释放、药效快等优势。mPEG-PLA聚合物是目前胶束给药系统中最有潜力的载体材料之一,不仅可以增加难溶性药物的溶解度,实现较大的载药量,而且具有良好的生物相容性以及可生物降解性,不会在体内积蓄而产生毒副作用,临床安全性高[32]。mPEG-PLA形成的胶束释放药物的速度较快,并且胶束主要在肝脏进行代谢,可以将氯吡格雷携带至肝脏,经肝药酶进行代谢,产生活性代谢产物,实现快速的抗血小板聚集的作用,满足临床急救的需求,有望为氯吡格雷注射剂的研制提供一种全新方案。
3 小结
在面临ACS的情况时,对患者给予氯吡格雷治疗的最佳剂量和时机目前尚无定论且备受争议。通常,随着氯吡格雷片剂的给药量增大,达到预期治疗作用的时间会缩短,但是过高的剂量会增加氯吡格雷的副作用,因此,在提高抗血小板效果的同时需要权衡因高剂量带来的出血风险。对于ACS患者,临床医生需决定其应在PCI前开始氯吡格雷负荷剂量的治疗,还是将治疗推迟到PCI术后。如果较早开始治疗,潜在缺血事件发生的风险可能会降低,可以避免患者出现梗死或再狭窄的情况;但如果血管造影显示需进行冠状动脉搭桥手术,那么负荷剂量氯吡格雷产生的抗血小板作用会使此手术方案复杂化,需推迟手术时间。然而,对于ACS,推迟手术可能严重威胁患者的生命安全。因此,开发快速起效的氯吡格雷注射剂型,可以有效填补ACS急诊手术用药方面的空白,其临床价值显而易见,具有广阔的开发前景。
多年来,全球范围内对氯吡格雷注射剂的研究十分活跃,本文所综述的研究为氯吡格雷注射剂型的开发提供了丰富的研究案例。但是,目前国内外尚无氯吡格雷注射剂型获准上市,大部分研究还处于临床前或临床Ⅰ期阶段,制备稳定性良好和副作用低的氯吡格雷的静脉注射剂仍面临较大挑战。随着相关研究的继续深入进行,新技术和新方法的日臻完善,相信氯吡格雷注射剂的开发会迎来新的突破。
