373例国外医疗器械召回事件分析及启示
2022-02-23赵玉娟田月洁
赵玉娟,赵 燕,李 倩,黄 琳,玄 怡,田月洁*
(1.山东省药品不良反应监测中心,济南 250014;2.国家药品监督管理局药品评价中心,北京 100022;3.潍坊市药品不良反应监测中心,山东潍坊 261000)
0 引言
医疗器械种类多、学科跨度大,近年来随着医疗服务的信息化、网络化,市场对高精尖医疗器械的需求逐渐增长,这使得医疗器械的风险管理也越来越重要。风险管理应该贯穿于医疗器械管理的全生命周期,包括产品研发、生产、销售、使用全过程[1]。通过开展上市后医疗器械不良事件监测等工作,对缺陷医疗器械产品实施召回是国际通行的监管手段,目的是确保产品使用的安全性并消除缺陷产品对公共健康安全的威胁[2]。美国是世界上最早实行产品召回的国家,1976年出台的《食品药品和化妆品法案》对医疗器械召回的规定也非常具体[3]。我国于2011年出台《医疗器械召回管理办法(试行)》,2017年进行修订并实施至今。据统计,我国近几年医疗器械召回数量不断增加,2010—2018年由我国国家药品监督管理局官网[4]公布的医疗器械召回事件共计1 533例,而2020年1 a就已达到492例,但分析这些数据发现,国内企业召回数量占比较小,召回总数量与召回工作较成熟的美国等国家召回的数量相比还存在一定的差距。本文对2011—2020年美国、英国、加拿大和澳大利亚官方网站发布的与医疗器械自身关联性较大的召回事件进行分析讨论,以期为我国医疗器械上市后风险管理和上市前风险因素识别提供参考。
1 资料与方法
1.1 资料来源
搜集2011—2020年美国食品药品监督管理局(Food and Drug Administration,FDA)[5]、英国药物和保健产品管理局(Medicines and Healthcare Products Regulatory Agency,MHRA)[6]、加拿大卫生部(Health Canada)[7]、澳大利亚治疗产品管理局(Therapeutic Goods Administration,TGA)[8]等官方网站发布的373例与医疗器械自身关联性较大的召回事件。
1.2 方法
对373例医疗器械召回事件从产品分类、召回原因归类及主要风险产品召回原因等方面进行分类统计分析,探讨其内在规律和事件特点。产品分类依据国家药品监督管理局发布的《医疗器械分类目录(2018年版)》[9],召回原因参照YY/T 0316—2016[10]中列出的与医疗器械有关的危险源进行归类分析,同时对召回事件具体表现和主要风险产品召回原因进行详细描述。
2 结果
2.1 产品分类
依据国家药品监督管理局发布的《医疗器械分类目录(2018年版)》的分类标准,373例召回事件产品涉及除中医器械外的其他21个子目录,详见表1。由表1可知,注输、护理和防护器械,呼吸、麻醉和急救器械最多,共计135例,占比为36.2%;其次是无源植入器械、临床检验器械、神经和心血管手术器械等。373例召回事件中,有7例涉及的是体外诊断试剂。
2.2 召回原因归类
参照YY/T 0316—2016中列出的与医疗器械有关的危险源的分类方法,将危险源分为能量危险源、生物化学危险源、操作危险源和信息危险源四大类,召回事件涉及12个小类,结合上市后医疗器械在使用过程中发生的其他情况,对召回原因进行归类分析,详见表2。
373例召回事件中,操作是主要的危险源,占59.5%。从分类上看,功能问题最多,有207例,占55.5%,主要是软件版本或软件系统问题、电池问题、不正确的测量结果和部件故障导致的功能失效或变坏,功能失效中尤其是报警功能失效较为突出,另外还有泄漏等原因造成的不正确输出,部件分离或不匹配造成无法使用等;其他分类共有146例,占39.1%,主要原因来自于机械能、电磁能、生物学的、化学的、使用错误和标记等。另外,还有20例无法用这12类危险源进行汇总,主要原因有不符合标准、装配错误、假冒产品等。
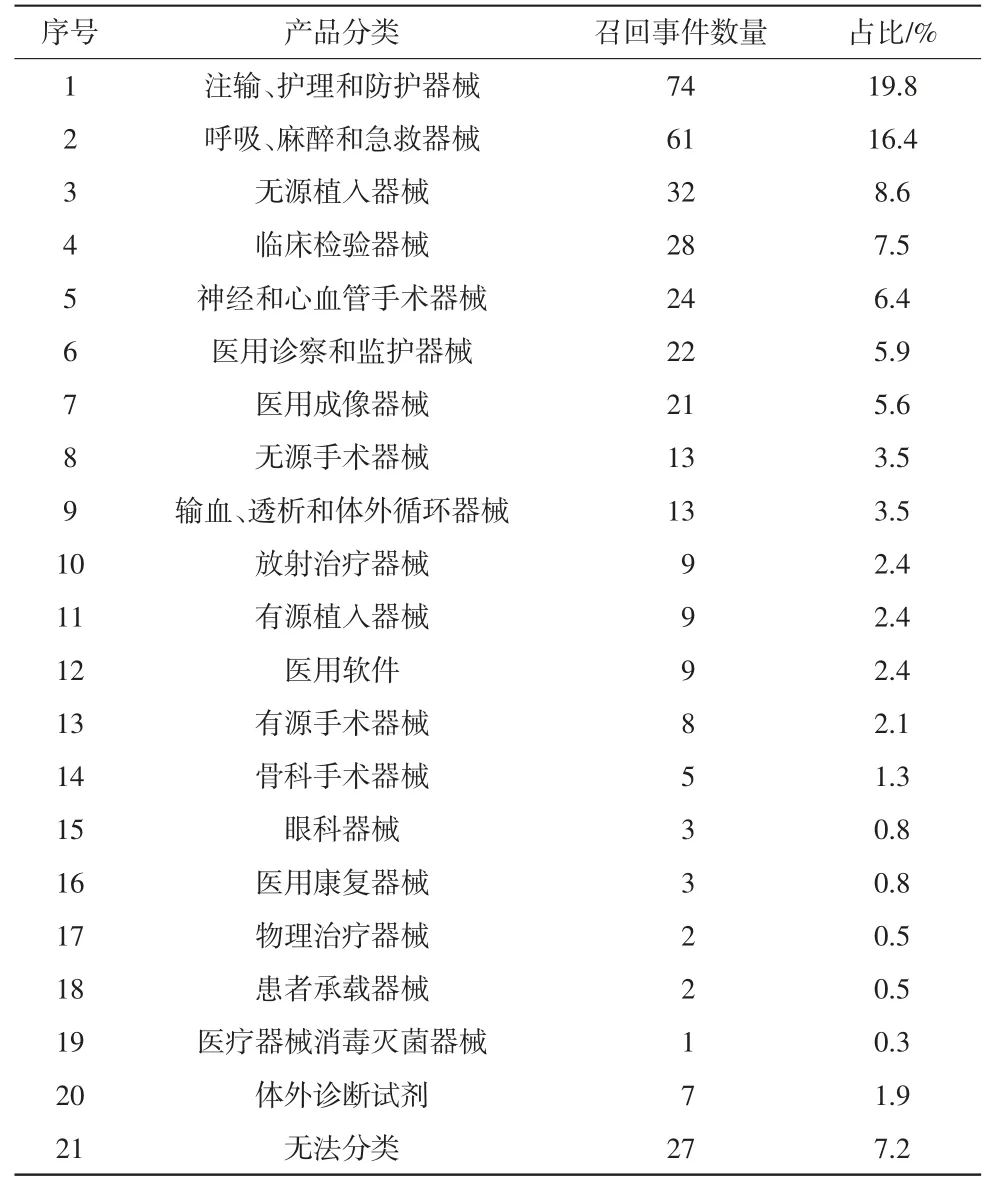
表1 召回事件涉及产品分类情况

表2 召回原因归类分析情况
2.3 主要风险产品召回原因
骨科、心血管医疗器械等高风险产品一直都是召回事件关注的重点[11-13],风险较高且召回数量较多的产品主要集中在数量前五位的产品分类中,具体统计分析情况见表3。

表3 主要风险产品召回原因分析
3 讨论
通过对373例召回事件进行分析,发现注输、护理和防护器械,呼吸、麻醉和急救器械风险高,召回事件也相对较多,尤其是这2个产品分类中的输注泵、注射器、输液器、呼吸机、自动除颤器、麻醉机等产品;其次是无源植入器械、临床检验器械、神经和心血管手术器械等,如骨科植入物、自动分析仪、血糖仪、导丝等。上述分析说明风险较高的医疗器械产品较容易发生召回。召回工作是不良事件监测风险的重要控制措施,根据国家药品不良反应监测中心发布的《国家医疗器械不良事件监测年度报告(2020年)》[14],报告数量前五位的产品是注输、护理和防护器械,医用诊察和监护器械,物理治疗器械,临床检验器械,呼吸、麻醉和急救器械,与英美国家召回事件数量较多的产品基本一致,说明不良事件较多的产品更容易发生召回,不同国家召回产品都是类似的高风险产品。但是也可以看到,召回数量较多的无源植入器械、神经和心血管手术器械等高风险产品的不良事件报告数量并不多,一方面与该类产品使用中影响因素多、不良事件发生原因复杂有关,另一方面也受使用单位对不良事件的理解和报告意识所影响。
医疗器械在真实世界环境中的使用情况可以更好地体现医疗器械的受益-风险[15]。美国FDA发布的《医疗器械上市前批准(PMA)和分类界定中风险受益评估考量因素指导原则》[16]提出,可将上市后的数据应用于受益-风险判定。Liebel等[17]也曾分析和使用美国制造商和用户设备使用体验(Manufacturer and User Facility Device Experience,MAUDE)数据库和医疗设备召回数据库系统,并建议制造商使用这2个数据库以识别未知的使用问题,发现设计需求并完善风险管理文件。医疗器械在上市前因科学认知有局限,临床试验时间短、范围窄等,很多问题无法及时发现[18]。通过同类产品召回事件追溯危险源,可以在设计研发阶段将这些风险因素考虑进去,避免该类事件再次发生。例如,骨科植入物的金属毒性是多例召回事件的重要原因,在设计研发同类产品时,应该着重考虑选材的金属特性、耐磨度等因素;有源设备类应关注功能失效问题,优化软件,提高功能易失效部件的品质等。
2018年我国发布新修订的《医疗器械不良事件监测和再评价管理办法》[19],要求医疗器械上市许可持有人对上市后医疗器械的安全性开展持续性研究。目前由企业提交的医疗器械不良事件报告所占比例仍然非常小[14],而且存在意识不强、调查评价能力有待提高等问题,部分企业上市后安全性监测方案过于简单,使得上市后收集的信息无法得到充分利用。2017年发布的《医疗器械召回管理办法》[20],明确了企业在召回工作中的主体责任,随后国内企业召回事件逐渐增多,但从数量和风险程度上与国外企业相比还存在较大差距。对比欧美国家的政策措施,依据欧盟《医疗器械法规》[21],制造商应在产品上市前制定上市后的监督计划,其中包含系统地收集相关数据信息,确定分析所用指标和阈值等,这就需要制造商在上市前研究同类产品的风险情况以确定风险触发阈值。美国已建立较为完善的医疗事故责任追溯制,若是因医疗器械潜在风险引起的事故,生产企业需要支付的赔偿金额要远高于事故之前通过自查和主动召回而消除潜在风险的成本。参考国外的召回事件,总结风险较高的产品及可能存在的缺陷项,可为企业开展主动召回工作提供思路,督促企业履职尽责。
4 结语
本文对2011—2020年美国等国家的药品监督管理部门官方网站发布的与医疗器械自身关联性较大的召回事件进行统计分析和讨论,结果表明风险高的产品更容易发生召回,同一类产品召回原因具有相似性。针对目前我国主动召回的现状,建议如下:(1)生产企业应树立全生命周期风险管理的理念,在自身产品特点的基础上,参考同类产品召回信息,制订上市后安全性监测方案,并将方案做细做实,尤其是关注召回等纠正预防措施的触发阈值、掌握各类产品可能存在的缺陷项及其风险控制措施;正确对待医疗器械不良事件报告,提高召回意识,合理利用收集的信息,为同类产品上市前的研发和改进工作提供参考;加强和医院的沟通,不仅对医务人员使用产品进行培训,还应充分告知产品可能存在的风险问题,提醒临床注意并及时反馈。(2)医疗机构应关注召回、警戒等信息,利用国家药品监督管理局网站中医疗器械召回、不良事件反馈等模块获知国内医疗器械风险处置情况,也可以登录其他国家药品监督管理部门网站了解国外医疗器械风险处置情况、相关案例和对召回事件的分析结论,用来提醒医务人员在临床使用时关注对应医疗器械的风险点;树立报告意识和风险意识,正确对待院内出现的医疗器械不良事件,对于企业改进或召回的产品及时跟踪处理,避免因不重视导致伤害事故的发生。召回是消除缺陷的一种行为,可以避免不必要的伤害,做好这项工作需要监管部门、各级医疗机构和医疗器械生产企业的共同努力和公众的配合。作为医疗器械全生命周期管理的一个重要环节,召回工作应该被正确对待,使之为民众的用械安全保驾护航。
