玉米开花期转录因子候选基因的关联分析
2022-02-22马拴红万炯梁瑞清张雪海邱小倩孟淑君徐宁坤林源党昆泰王琪月赵嘉雯丁冬汤继华
马拴红,万炯,梁瑞清,张雪海,邱小倩,孟淑君,徐宁坤,林源,党昆泰,王琪月,赵嘉雯,丁冬,汤继华
玉米开花期转录因子候选基因的关联分析
马拴红1,万炯1,梁瑞清2,张雪海1,邱小倩1,孟淑君1,徐宁坤1,林源1,党昆泰1,王琪月1,赵嘉雯1,丁冬1,汤继华1
1河南农业大学农学院/省部共建小麦玉米作物学国家重点实验室,郑州 450002;2华南农业大学农学院,广州 510642
【】生育期相关性状是玉米育种研究的重点之一。作为重要的生育期性状,开花期(抽穗期、吐丝期和散粉期)的提前,可保证玉米充分脱水,适宜机收;也为中国黄淮海地区小麦-玉米一年两熟耕作模式下的小麦播种减轻压力。转录因子是转录水平基因表达调控的重要上游因子,对目标基因发挥转录激活或转录抑制的作用。在全基因组水平上解析转录因子对玉米开花期的调控作用,获得开花期提前且不影响产量的玉米转录因子单倍型组合,进而挖掘优异种质资源,可为培育开花期适宜的玉米育种研究提供基因资源。使用候选基因关联分析方法,对开花期相关转录因子及显著SNP进行分析;并利用DAP-seq技术捕获了关键转录因子的结合位点和下游基因;随后对转录因子调控的下游基因进行GO分析,探究转录因子通过影响其下游基因对开花期进行调控的基因网络。玉米开花期3种性状(吐丝期、散粉期、抽穗期)中,与吐丝期和抽穗期性状以及吐丝期和散粉期性状同时关联的转录因子显著SNP均为75个,与抽穗期和散粉期性状同时关联的显著SNP为128个,同时关联到3种表型的显著SNP为58个。表明开花期3种性状可能受相同的转录因子调控。选取含有3个及以上开花期相关显著SNP的转录因子基因,通过DAP-seq,捕获了这些转录因子结合的关键基序及调控的下游基因。转录因子结合的下游基因显著富集于转录因子活性、DNA结合、RNA结合、有机氮化合物的合成代谢过程、与生殖有关的发育过程等;不同的转录因子存在共同调控的下游基因,生育期相关性状关键调控性转录因子为ARF、MYB和NAC。对关键上游转录因子进行单倍型分析,发掘了玉米生育期提前,同时对产量无负向影响的转录因子最优单倍型组合。运用DAP-seq技术并结合前人研究绘制了全基因组水平上转录因子对生育期相关农艺性状的调控网络,并发掘了既提前玉米生育期又对产量无负向影响的转录因子最优单倍型组合。
玉米;转录因子;开花期;DAP-seq;单倍型
0 引言
【研究意义】作为一年两熟耕作制度的重要一环,玉米开花期(抽穗期、吐丝期和散粉期)的长短直接影响黄淮海区域小麦正常播期[1]。玉米开花期的提前,也使玉米籽粒充分脱水,适宜机械化直收籽粒[2]。转录因子是转录水平基因表达调控的重要上游因子,对目标基因发挥转录激活或转录抑制的作用[3-4]。解析转录因子对玉米开花期的调控作用,获得开花期提前且不影响产量的玉米品种,不仅可以加速实现玉米的机械化收获,也可以在玉米-小麦的整体水平上提高粮食产量,有效保障中国的粮食安全。【前人研究进展】开花期相关性状作为一年两熟耕作模式的制约因素,是玉米育种研究的重点之一。Guo等[5]通过关联分析发现,在玉米成花基因启动子区域存在一个与开花时间显著关联的SNP(SNP-1245)。Huang等[6]通过图位克隆将开花期数量性状基因定位在上游57 kb的Harbinger-like转座子上,该基因负调节成花基因的表达,导致长日照条件下开花延迟。也是开花抑制因子,该基因的过表达将延迟玉米花期[7]。在基因表达过程中,转录的起始是最为重要的调控步骤[3]。转录因子(transcription factors,TFs)是转录水平基因表达调控的重要上游因子,通过与目标基因启动子区的顺式作用位点(-acting elements)相结合,对目标基因发挥转录激活或转录抑制的作用[4]。MYB转录因子家族规模庞大,参与控制生物和非生物胁迫反应、发育等多种过程,已经在拟南芥、水稻、烟草、棉花、辣椒等多个物种中被报道为花器官相关调控转录因子[8-13]。WRKY转录因子几乎只在植物中发现[14],自最初从甘薯中分离出以来[15],已从几种高等植物中鉴定出大量WRKY转录因子[16-19]。在拟南芥中,WRKY12和WRKY13多次被报道为开花期相关转录因子[20-21]。而其他转录因子,如PIF与VOZ也被报道为开花期相关转录因子[22-23]。此外,转录因子OsLFL1的过表达延迟了水稻的开花时间[24];转录因子ZmMADS1被认为是玉米开花时间调控因子[25];番茄中转录因子SlZFP2[26]和菠萝中转录因子bHLH2(AcCIB2)也参与开花时间调节[27]。DNA亲和纯化测序(DNA affinity purification sequencing,DAP-seq)是一种高通量、高分辨率的技术,用于鉴定全基因组转录因子结合位点,已成为解析基因表达调控网络的有力工具[28]。由于DAP-seq使用外源表达的转录因子(与Halo标签形成融合蛋白)直接捕获基因组DNA,不需要标记的转基因系或基因特异性抗体,同时仍然可在全基因组序列中捕获转录因子结合事件,越来越多的研究人员基于该技术开展研究[29-32]。【本研究切入点】目前,已有研究报道转录因子对表型的调控作用,然而,在全基因组解析转录因子对表型调控,尤其是对开花期等重要生育期相关表型调控的研究鲜见报道。【拟解决的关键问题】本研究以玉米开花期(抽穗期、吐丝期和散粉期)性状作为切入点,筛选其相关候选转录因子,利用DAP-seq技术探究抽穗期、吐丝期和散粉期性状相关关键转录因子的结合序列和下游基因,同时进行优异单倍型分析,为培育开花期适宜的玉米育种研究提供基因资源。
1 材料与方法
1.1 试验材料
选取20粒籽粒饱满无破损的B73种子,于2020年在室内种植于营养土内,长至两叶一心后,转移至暗培养,生长至三叶期,收集黄化叶片用于DAP-seq试验中DNA的提取。
1.2 DAP-seq流程
根据前期研究,选取转录因子内包含3个及以上显著性SNP的MYB、NAC、ARF等25个家族的40个转录因子[33],利用TnT® SP6 High-Yield Master Mix Minus Amino Acids(Promega,美国)无细胞表达试剂盒体外表达DAP-seq试验所需的转录因子蛋白。用CTAB法提取玉米B73叶片DNA,并使用微波破碎仪M220(Covaris,美国)破碎约至200 bp。对破碎后的片段进行纯化,去除过大或者过小的片段。取5 μg纯化后的片段DNA,利用NEXTflex™ Rapid DNA-Seq Kit建库试剂盒(Bio Scientific,美国)对片段DNA末端加poly(A),以构建DAP DNA文库。将体外表达的融合蛋白与Magne HaloTag Beads(Promege,美国)一起孵育,通过Beads捕获标签的方式固定转录因子,与构建好的DAP DNA文库进行反应[34]。随后捕获并洗脱与融合蛋白特异结合的DNA片段,将DNA片段加上高通量测序用的长接头(各转录因子使用特异的index接头)进行测序。构建好的文库送至贝瑞基因(贝瑞和康生物技术有限公司,北京),采用Illumina Novaseq对40个文库进行双末端混合测序。根据特异index对测序数据进行拆分,并提供拆分后数据作为测序原始数据。
1.3 DAP-seq数据分析
得到原始数据后,使用Trimmomatic软件(https://github.com/usadellab/Trimmomatic)对数据进行质控和筛选,以Q30为质控标准,去除含有接头序列的reads、N比例大于10%的reads以及低质量reads。使用Bowtie2软件(https://github.com/BenLangmead/bowtie2)将Clean data比对到B73 RefGen_v4(ftp://ftp. ensemblgenomes.org/pub/release-50/plants/fasta/zea_mays/ dna/Zea_mays.B73_RefGen_v4.dna.toplevel.fa.gz)参考基因组。使用空白HALO标签(未融合转录因子)与DAP DNA文库结合的测序文件作为空白对照,使用MACS2软件(https://hbctraining.github.io/Intro-to- ChIPseq/lessons/05_peak_calling_macs.html)对每个转录因子的结合片段分别进行结合峰的捕捉。并利用R软件的ChIPseeker包(https:// bioconductor.org/packages/ release/bioc/html/ChIPseeker.html)对富集的结合峰进行注释。根据结合峰最高点上下游200 bp的物理位置,使用Bedtools软件(https://bedtools. readthedocs.io/en/ latest/index.html)提取峰上下游200 bp的序列,使用MEME-ChIP软件(https://meme-suite.org/meme/tools/ meme-chip)对每一个转录因子分别进行保守motif分析。筛选了位于基因启动子区域(转录起始位点上游2 000 bp)的结合峰,并使用植物基因功能分类数据库AgriGO对这些基因进行GO富集分析(http://systemsbiology.cau.edu.cn/agriGOv2),使用R软件ggplot2包(https://ggplot2.tidyverse.org/)进行数据结果可视化。
1.4 开花期相关转录因子单倍型分析
候选基因关联分析所用到的重测序数据[35]和21个表型数据[36-37]均由华中农业大学作物遗传改良国家重点实验室严建兵教授提供。根据候选基因关联分析和DAP-seq分析结果,本研究筛选、和3个基因中和表型性状相关的显著SNP。根据该位点不同的SNP类型,将关联群体划分为不同的单倍型并和表型值一一对应。根据表型值,通过单因素方差分析,以<0.01为阈值检验单倍型之间的差异。利用R语言绘制单倍型图。
2 结果
2.1 开花期相关性状显著性SNP和显著性转录因子
开花期性状是玉米生长发育的重要性状。抽穗期、吐丝期和散粉期不仅通过影响玉米的正常授粉而影响玉米产量,同时,缩短玉米生长期,培育生育期适宜玉米的关键之一。前期研究中,对玉米关联群体的21个农艺及产量性状进行了全基因组关联分析[36-37],并结合全基因组范围内的81个转录因子家族的2 034个转录因子序列,进行了候选基因关联分析[33]。本研究在此基础上,进一步分析开花期相关性状显著性SNP和显著性转录因子。
抽穗期、吐丝期和散粉期3种性状共关联到122个转录因子,涉及551个显著SNP。在这三种性状中,与吐丝期和抽穗期性状以及吐丝期和散粉期性状同时关联的显著SNP均为75个,与抽穗期和散粉期表型同时关联的显著SNP为128个,同时关联到3种表型性状的显著SNP为58个(表1)。表明开花期3种性状可能受相同的转录因子调控。
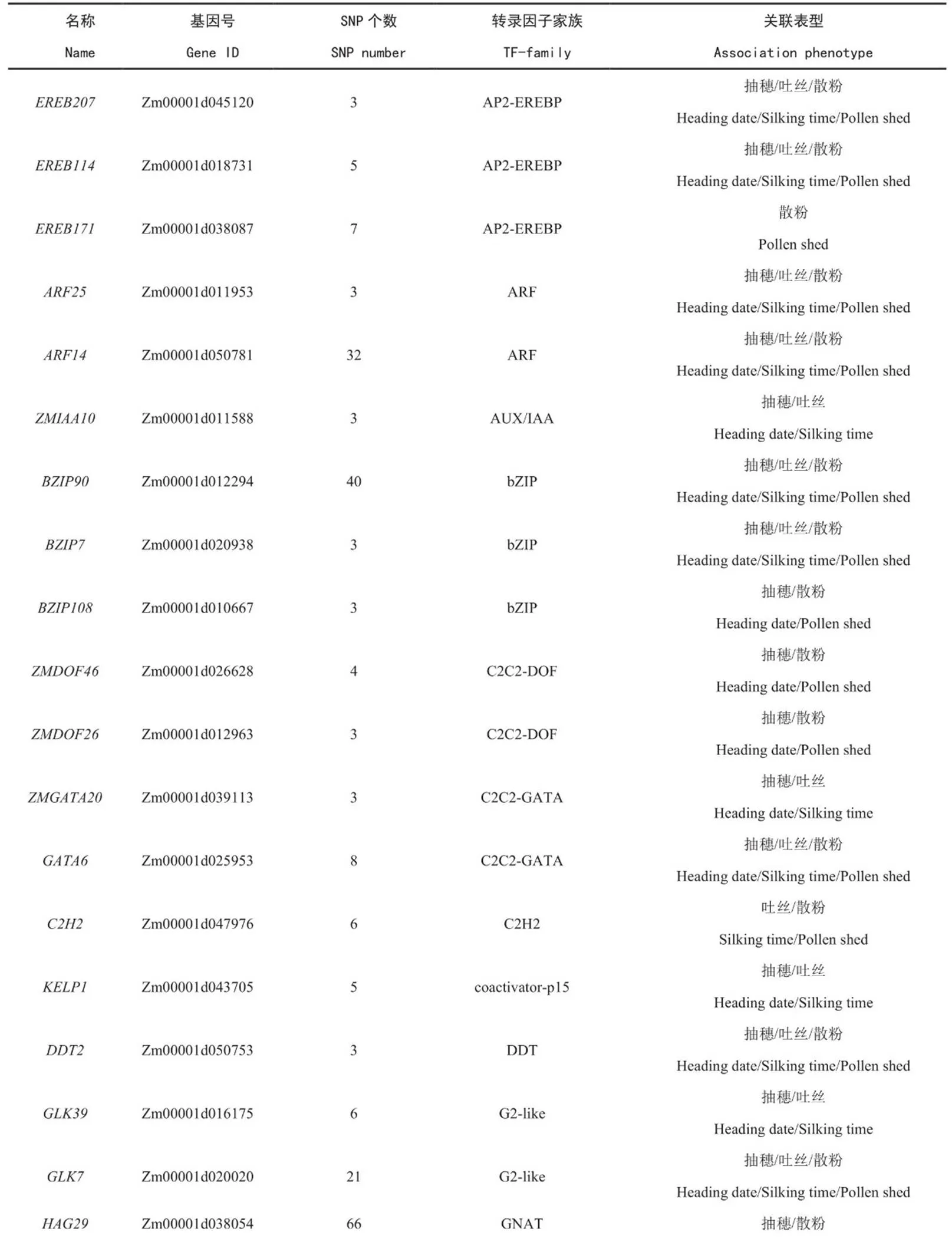
开花期相关转录因子包含的显著SNP数量从1到70不等。将转录因子按基因内包含显著SNP数目进行分类,包含2个及以上显著SNP的转录因子有70个;包含3个及以上显著SNP转录因子有47个;包含5个及以上显著SNP转录因子有20个。选取转录因子内包含3个及以上显著SNP的转录因子共40个(电子附表1)进行了DAP-seq分析,以捕获其下游基因。

表1 开花期性状相关显著的SNP数目及转录因子统计
抽穗/吐丝:抽穗期和吐丝期表型共同关联到的SNP、转录因子等;抽穗/散粉:抽穗期和散粉期表型共同关联到的SNP、转录因子等;吐丝/散粉:吐丝期和散粉期表型共同关联到的SNP、转录因子等;抽穗/吐丝/散粉:抽穗期、吐丝期和散粉期表型共同关联到的SNP、转录因子等
Heading date/Silking time: SNPs and TFs associated with the phenotype at both heading date and silking time; Heading date/Pollen shed: SNPs and TFs associated with the phenotypes at both heading date and pollen shed; Silking time/Pollen shed: SNPs and TFs associated with the phenotypes at both silking time and pollen shed;Heading/silking/loose powder: SNPs and TFs associated with all the phenotypes of heading date, silking time and pollen shed
2.2 转录因子结合峰在全基因组的分布
通过高通量测序,获得DAP-seq原始数据。参考B73 RefGen_v4基因组序列,得到转录因子结合的peaks。对所有转录因子结合的peaks进行汇总和分类,总计513 960个peaks分布在基因间区,占总数目的74.49%;74 575个peaks分布在启动子区,占总数目的10.81%(图1-A)。对位于基因启动子区域的peaks进一步统计分析,其中51.59%的peaks位于转录起始位点(transcription starting site,TSS)上游500 bp范围内(图1-B)。鉴于转录因子不仅可以在基因启动子区域的近端发挥表达调控作用,也可在基因间区以及基因远端发挥作用[38],这些抽穗期、吐丝期和散粉期相关转录因子结合peaks的分布符合转录因子结合位点的分布规律。
2.3 开花期相关转录因子motif分析
对DAP-seq结果进行分析发现,有22个转录因子结合peaks数目超过1 000。为得到准确的转录因子的结合基序(motif)信息,对这22个转录因子进行了分析。22个转录因子分属于bZIP、HB、NAC、MYB、ARF、AP2-EREBP、C2C2-GATA、C2H2、G2-like、WRKY、GRF、PLATZ、NLP和MADS 14个转录因子家族。ARF25和ARF14结合的基序为TGTCGG;bZIP7和bZIP90结合的序列为TGACCTGA;NAC3结合的序列为CCCTNNNNNNNACGGC;NAC16结合的序列为CTTNNNNNNNAAGCT;NAC57结合的序列为CAAGCAA;NAC114结合的序列为TTGCGTGT。MYB36在2个重复中捕捉到相同的序列,其序列为TAACTGAC;而MYB23结合的序列与之略有差异,为CAACTAC。对HB家族的2个成员来说,HB62结合的序列为AATNATTA,而HB123结合的序列为ATCAATCA。EREBP207、C2H2、ZmGATA20、GLK39、WRKY117、GRF14、PLATZ9、NLP2和MADS73转录因子均仅检测到单一的家族成员,其结合的基序分别为GGCGGCGGCGGCG、TTTGTCTTTT、GATC、ATTCT、AAAGTCAAA、TGTCAG、TANAATT、AAACGTCATA和CCAAAAANGGAAA(图2)。

A:所有peaks在全基因组的分布;B:转录起始位点上游2 000 bp范围内的peaks的分布
2.4 转录因子调控下游基因分析
对单个转录因子进行注释,并筛选结合峰位于启动子区域的基因作为该转录因子调控的下游基因(电子附表2)。对这些转录因子下游基因进行GO(gene ontology)富集分析发现,这些下游基因显著富集在生殖发育、细胞分裂、植物器官发育等过程;在生物学过程中被显著富集的是有机氮化合物的合成代谢过程、减数分裂过程、与生殖有关的发育过程等;在分子功能中显著富集的是RNA结合、转录因子活性、序列特异性DNA结合、丝氨酸/苏氨酸蛋白激酶活性等(图3)。
对不同转录因子DAP-seq结合的下游基因进行了功能注释。结果显示,不同的转录因子可结合在共同的下游基因启动子区域,也许协同调控下游基因的转录。编码不依赖于光周期的早花蛋白,该基因启动子被ARF、MYB、HB和NAC等9个转录因子家族的成员共同调控。此外,、、等注释为早花蛋白基因,它们受到ARF、MYB和NAC转录因子的结合与调控。暗示开花期相关性状关键共性调控性转录因子为ARF、MYB和NAC,它们通过影响早花蛋白基因等下游基因的表达控制玉米开花期(表2)。
2.5 单倍型分析
对ARF、MYB和NAC成员、和进行分析,结果显示,基因区域中相邻的SNP之间具有较强的相关性。由于3种开花期性状之间存在共同关联的显著SNP,对、和分别选取16、2和6个显著SNP进行单倍型分析。定义开花期提前的单倍型为优异单倍型,中,抽穗期和散粉期的优异单倍型为单倍型1,吐丝期的优异单倍型为单倍型2(图4-A);中,抽穗期和散粉期的优异单倍型均为单倍型1(图4-B);中,抽穗期、吐丝期和散粉期的优异单倍型均为单倍型3(图4-C)。为探究最优单倍型组合和产量的关系,将不同单倍型进行组合。A、B、C分别代表、和,1代表优异单倍型,2代表非优单倍型(表3)。与508份自交系的平均值相比,最优单倍型开花期3种表型均明显提前,而单穗产量也明显下降。在非优单倍型的简单组合(A2B2C2)中,开花期表型均无明显提前,而单穗产量也无明显变化;在最优单倍型的简单组合(A1B1C1)中,开花期表型均明显提前,而单穗产量却明显下降;相反,A1B2C2组合中,开花期表型均明显提前,而单穗产量也略有升高(表3)。因此,在育种过程中,为获得开花期与产量的平衡,可使用分子标记跟踪筛选具有A1B2C2基因型组合的玉米品系。

图2 不同转录因子DAP-seq结合的motif
3 讨论
3.1 不同作物之间转录因子结合序列的保守性
转录因子结合基序(Motif)是一段典型的序列或者结构,是包含序列特异性的结合位点,或者是涉及某一个特定生物学过程的共性序列区段。基于motif序列的提取,可以预测潜在的结合位点等,有助于进一步理解各生物学过程中涉及的蛋白质-DNA互作事件,进而解析基因的表达调控。通过对开花期相关转录因子开展DAP-seq,捕捉到了转录因子与下游基因的结合序列。为研究转录因子家族在不同作物之间的结合序列保守性,结合拟南芥已公布的motif信息(http://neomorph.salk.edu/dap_web/pages)与本研究捕获的玉米转录因子motif进行了比对分析。bZIP家族(TGACNTNA)、NAC家族(CTT(CGT)NNNNNNNACG(AAG))、MYB家族(NAACTNNC)、ARF家族(TGTCGG)、GLK家族(ATTCT)、GRF家族(TGTCAG)等家族成员在玉米中结合的motif序列均与拟南芥一致;PLATZ9(TANAATT)和NLP2(AAACGTCATA)序列和拟南芥中(GAANNTTC TAGA、CAGCA)相应motif不一致(图5)。以上结果说明,转录因子与下游的结合位点有些是进化保守的,如MYB、NAC等家族,少数转录因子家族则存在单/双子叶植物间的差别。
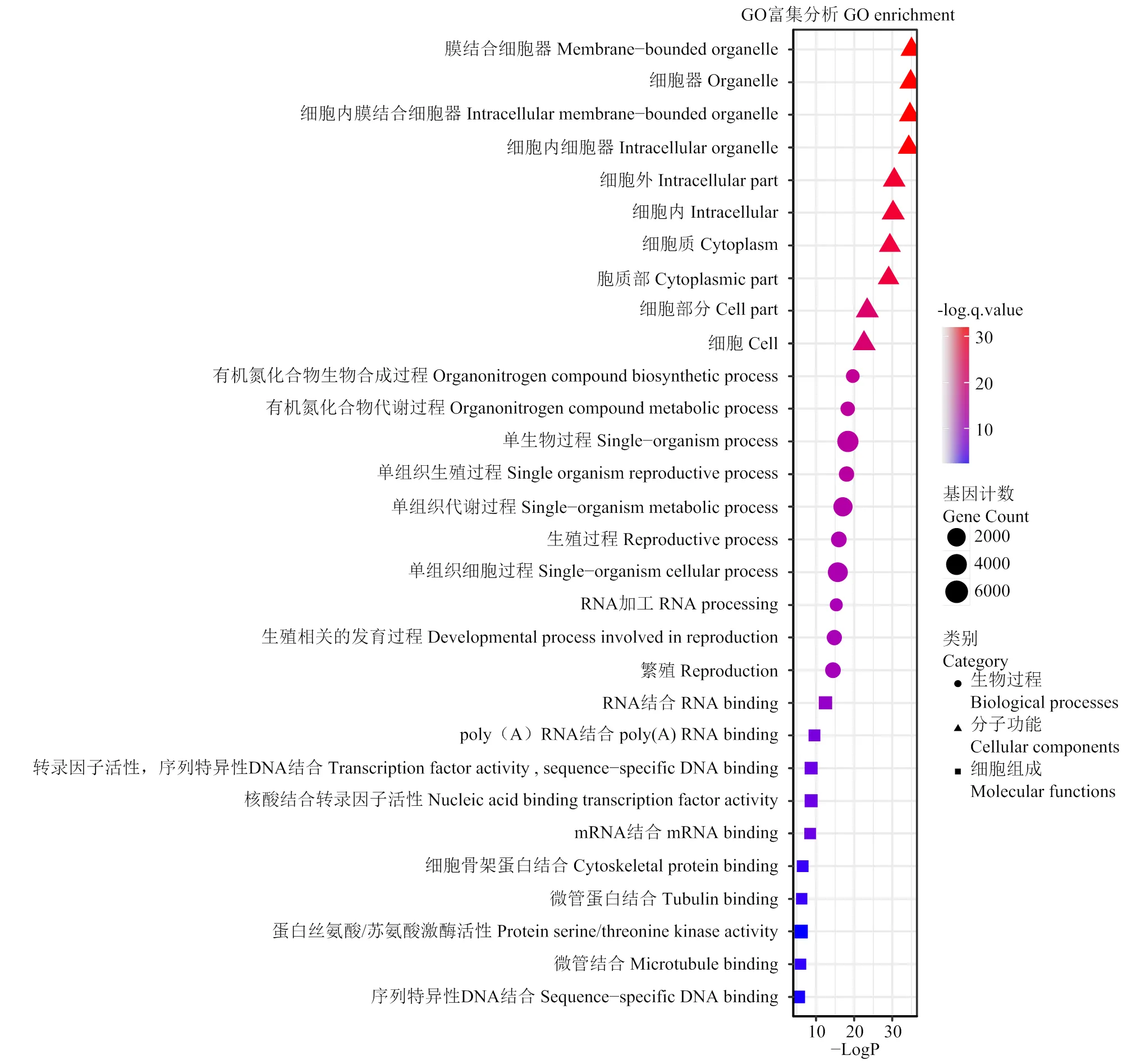
图3 DAP-seq结合下游基因的GO分析
3.2 转录因子及下游基因对开花期的调控
通过对DAP-seq捕捉到的下游基因进行GO分析,发现转录因子结合的下游基因富集于玉米生殖生长通路。编码光周期非依赖性早花基因在NAC、MYB、ARF等9个家族结合的下游基因中被发现。前人研究发现,光周期素依赖性早花1()的突变抑制了开花位点C(FLC)介导的开花延迟,突变导致非依赖性光周期中的早期开花,与FLC无关[39];编码多梳蛋白胚胎花2的基因在NAC和MYB家族结合的下游基因中被检测到。Bai等[40]发现拟南芥胚胎花1()基因的突变影响胚芽顶端发育,导致胚芽生长为花序,暗示了在调节营养生长向生殖生长转换中起作用。水稻编码一种假定的EMF蛋白,突变导致水稻植株提前开花[41]。此外,2个编码早花蛋白2()的基因也在多个转录因子家族(NAC、MYB、ARF)的下游基因中被检出。早花蛋白在拟南芥中多次被报道与昼夜节律光周期的调控相关,导致植株开花时间的改变[42-44]。在水稻中,OsELF3-1通过激活Ehd1的表达参与蓝光信号,促进短日照条件下的水稻开花;还抑制开花抑制子从而在长日照下间接促进开花[45]。在木薯中为晚间表达基因,该基因是4的直系同源基因,互补转基因可恢复拟南芥的生长习性,表现开花提前[46]。Huang等[47]发现二穗短柄草和狗尾草尽管在1.8亿年前已经分离,但其拟南芥同源基因和仍能在分子和生理水平上拯救拟南芥中的缺失。这些结果证明是在不同物种间保守的调控光周期和开花时间的功能基因。
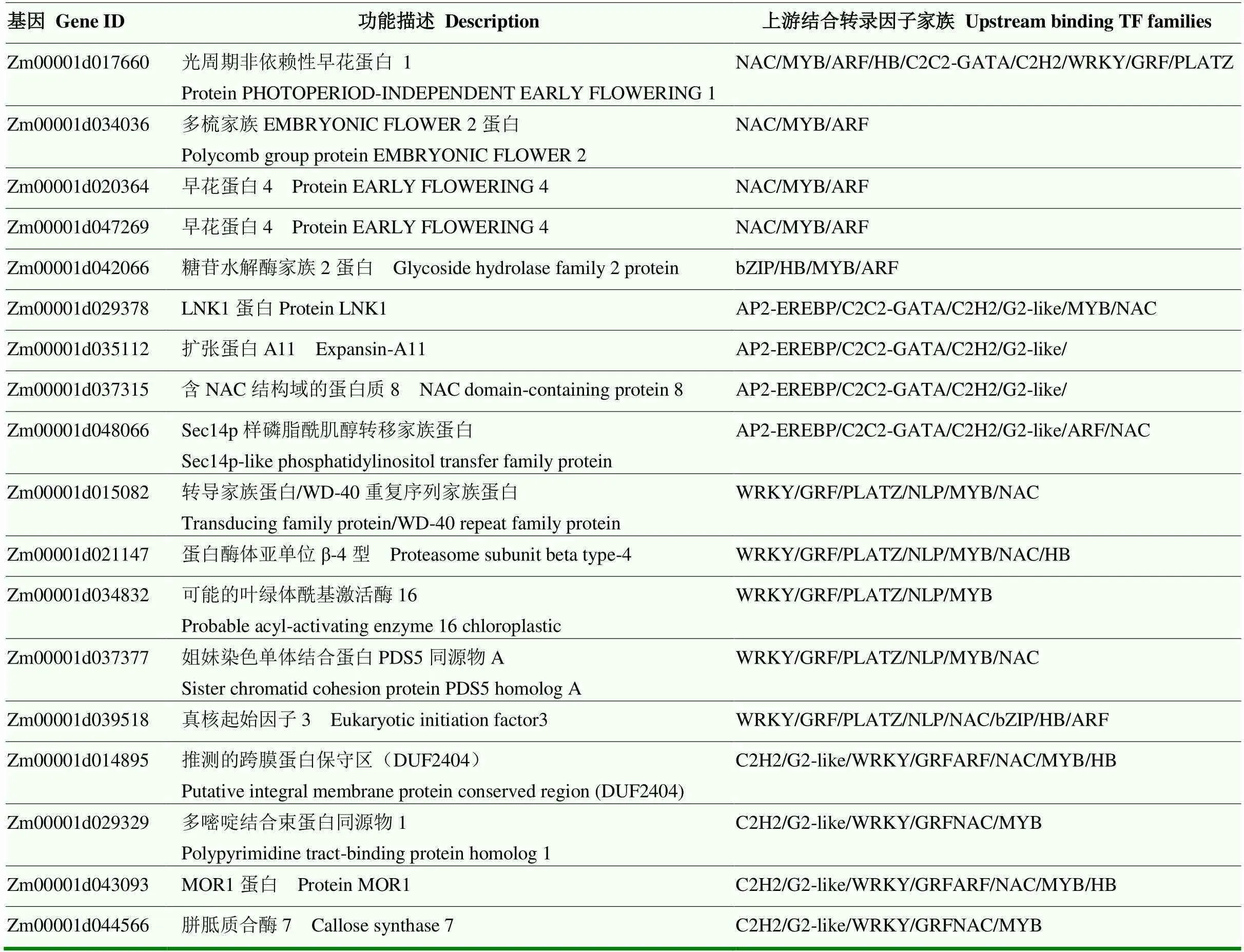
表2 转录因子调控的下游基因
编码LNK1蛋白的基因是AP2-EREBP、C2C2- GATA、C2H2、G2-like、MYB和NAC转录因子共同调控的下游基因。LNK缺乏已知的DNA结合域,然而,LNK1和LNK2作为辅助因子,通过与RVE4(REVEILLE)和RVE5相互作用与和()的启动子结合,从而调控光周期反应,控制开花时间[48]。胼胝质合成酶是控制胼胝质合成的关键酶,在植物生长发育和抗逆胁迫中具有重要作用[49]。胼胝质在多种作物中被报道与小孢子及花粉发育相关,胼胝质沉积异常,过多沉积或沉积量不足,提前降解或延迟降解都会导致花粉败育,从而造成植物雄性不育[50]。拟南芥中,ARF17能直接结合到胼胝质合酶基因CALLOSESYNTHASE5()启动子上,调控胼胝质合成和初生外壁形成,从而影响生长素信号途径调控的花粉壁和花药发育[51]。本研究中,胼胝质合酶基因是C2H2、G2-like、WRKY、GRF、NAC和MYB转录因子共同的下游基因。暗示转录因子可能通过与的启动子结合,调控玉米开花和花粉发育。基于开花期性状相关基因的功能,构建了依赖转录因子的玉米开花期性状相关基因表达调控网络(图6)。

A:ARF14;B:MYB64;C:NAC3。hap1—hap5:单倍型1—5 A: ARF14; B: MYB64; C: NAC3. hap1- hap5: haplotype 1-5

图5 拟南芥与玉米不同转录因子结合的motif
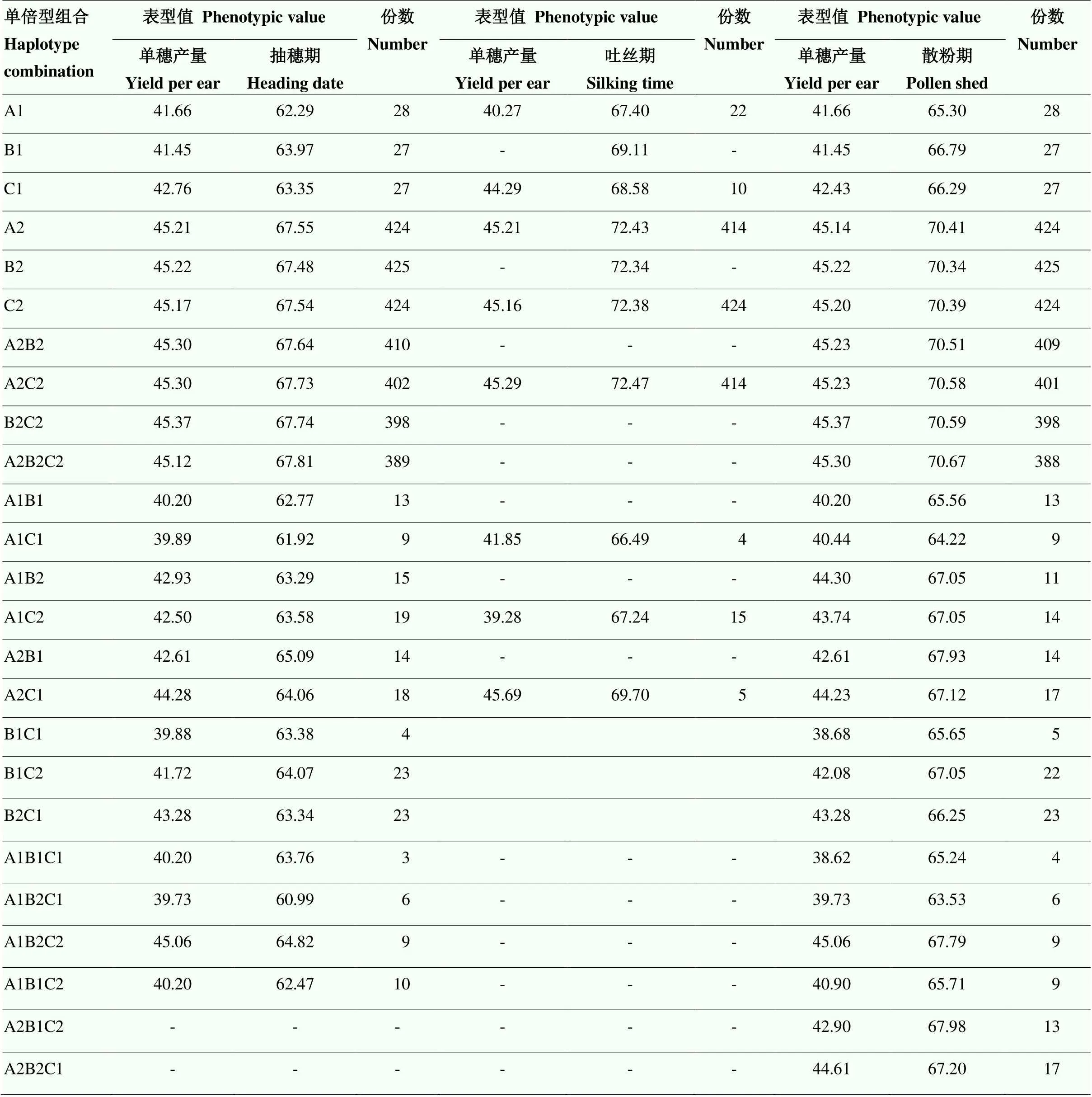
表3 关键转录因子不同单倍型组合对应的产量和开花期表型
A、B、C分别代表、和;508份自交系百粒重、抽穗期、吐丝期和散粉期平均值分别为44.74 g、67.34 d、70.20 d和72.18 d
A, B and C represent,andrespectively; The average values of 100 kernels weight, heading date, silking time and pollen shed of 508 inbred lines were 44.74 g, 67.34 d, 70.20 d and 72.18 d, respectively
3.3 优异单倍型在育种中的应用
作物种质资源中包含了大量的优异基因,发掘这些优异等位基因,可以鉴别与表型性状相关的候选基因核苷酸变异,以及优异单倍型的利用。本研究中,通过候选基因关联分析,获得大量和表型直接相关的SNP,且大多SNP高度连锁不平衡。对得到的显著SNP进行单倍型分析。结合产量数据,筛选出育种中可利用的优异单倍型组合(A1B2C2),证明在实际生产中可以实现既提前开花期,又对产量无负面影响的最佳田间表现;而根据这些单倍型开发的分子标记,可应用于追踪和预测目的性状的田间表现。

图中实线表示已有文献报道,虚线表示根据已有报道推测。黑色线条和红色线条表示不同的具体通路
4 结论
基于前期玉米转录因子候选基因关联分析结果进一步分析,发现开花期3种表型可能受相同的遗传调控,且开花期相关性状关键调控性转录因子为ARF、MYB和NAC;最优单倍型组合抽穗期和散粉期的表型值分别提前4.87和6.47 d,其对应的单穗产量则提高0.32 g,可以通过转录因子的优异单倍型组合获得最佳田间表现。
[1] 周宝元, 马玮, 孙雪芳, 高卓晗, 丁在松, 李从锋, 赵明. 播/收期对冬小麦-夏玉米一年两熟模式周年气候资源分配与利用特征的影响. 中国农业科学, 2019, 52(9): 1501-1517.
ZHOU B Y, MA W, SUN X F, GAO Z H, DING Z S, LI C F, ZHAO M. Effects of different sowing and harvest dates of winter wheat-summer maize under double cropping system on the annual climate resource distribution and utilization. Scientia Agricultura Sinica, 2019, 52(9): 1501-1517. (in Chinese)
[2] 柴宗文, 王克如, 郭银巧, 谢瑞芝, 李璐璐, 明博, 侯鹏, 刘朝巍, 初振东, 张万旭, 张国强, 刘广周, 李少昆. 玉米机械粒收质量现状及其与含水率的关系. 中国农业科学, 2017, 50(11): 2036-2043.
CHAI Z W, WANG K R, GUO Y Q, XIE R Z, LI L L, MING B, HOU P, LIU C W, CHU Z D, ZHANG W X, ZHANG G Q, LIU G Z, LI S K. Current status of maize mechanical grain harvesting and its relationship with grain moisture content. Scientia Agricultura Sinica, 2017, 50(11): 2036-2043. (in Chinese)
[3] MENG C A, FAZAL F M, BLOCK S M. Real-time observation of polymerase-promoter contact remodeling during transcription initiation. Nature communications, 2017, 8(1): 1-9.
[4] YANAGISAWA S. Transcription factors in plants: physiological functions and regulation of expression. Journal of Plant Research, 1998, 111(3): 363-371.
[5] GUO L, WANG X, ZHAO M, HUANG C, LI C, LI D, YANG C J, YORK A M, XUE W, XU G H, LIANG Y M, CHEN Q Y, DOEBLEY J F, TIAN F. Stepwise cis-regulatory changes in ZCN8 contribute to maize flowering-time adaptation. Current biology, 2018, 28(18): 3005-3015.
[6] HUANG C, SUN H, XU D, CHEN Q, LIANG Y M, WANG X F, XU G H, TIAN J G, WANG C L, LI D, WU L S, YANG X H, JIN W W, DOEBLEY J F, TIAN F. ZmCCT9 enhances maize adaptation to higher latitudes. Proceedings of the National Academy of Sciences of the USA, 2018, 115(2): E334-E341.
[7] STEPHENSON E, ESTRADA S, MENG X, OURADA J, MUSZYNSKI M G, HABBEN J E, DANILEVSKAYAET O N. Over-expression of the photoperiod response regulator ZmCCT10 modifies plant architecture, flowering time and inflorescence morphology in maize. PloS one, 2019, 14(2): e0203728.
[8] LI Y, JIANG J, DU M L, LI L, WANG X L, LI X B. A cotton gene encoding MYB-like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant and cell physiology, 2013, 54(6): 893-906.
[9] SHEN X P, HU Z W, XIANG X, XU L A, CAO J S. Overexpression of a stamen-specific R2R3-MYB gene BcMF28 causes aberrant stamen development in transgenic. Biochemical and biophysical research communications, 2019, 518(4): 726-731.
[10] AYA K, UEGUCHI-TANAKA M, KONDO M, HAMADA K, YANO K, NISHIMURA M, MATSUOKA M. Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. The Plant Cell, 2009, 21(5): 1453-1472.
[11] RAHIM M A, RESENTINI F, DALLA VECCHIA F, TRAINOTTI L. Effects on plant growth and reproduction of a peach R2R3-MYB transcription factor overexpressed in tobacco. Frontiers in plant science, 2019, 10: 1143.
[12] SUN B M, ZHU Z S, CHEN C J, CHEN G J, CAO B H, CHEN C M, LEI J J. Jasmonate-inducible R2R3-MYB transcription factor regulates capsaicinoid biosynthesis and stamen development in Capsicum. Journal of agricultural and food chemistry, 2019, 67(39): 10891-10903.
[13] HU R, YUAN C, NIU Y, TANG Q, WEI D, WANG Z. Regulation of plant MYB transcription factors in anther development. Chinese Journal of Biotechnology, 2020, 36(11): 2277-2286.
[14] LI S J, ZHOU X, CHEN L G, HUANG W D, YU D Q. Functional characterization ofWRKY39 in heat stress. Molecules and cells, 2010, 29(5): 475-483.
[15] ISHIGURO S, NAKAMURA K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Molecular and General Genetics, 1994, 244(6): 563-571.
[16] ÜLKER B, SOMSSICH I E. WRKY transcription factors: from DNA binding towards biological function. Current opinion in plant biology, 2004, 7(5): 491-498.
[17] PANDEY S P, SOMSSICH I E. The role of WRKY transcription factors in plant immunity. Plant physiology, 2009, 150(4): 1648-1655.
[18] WEI K F, CHEN J, CHEN Y F, WU L J, XIE D X. Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA research, 2012, 19(2): 153-164.
[19] RUSHTON D L, TRIPATHI P, RABARA R C, LIN J, RINGLER P, BOKEN A K, LANGUM T J, SMIDT L, BOOMSMA D D, EMME N J, CHEN X F, FINER J J, SHEN Q J, RUSHTON P J. WRKY transcription factors: key components in abscisic acid signalling. Plant biotechnology journal, 2012, 10(1): 2-11.
[20] MA Z, LI W, WANG H, YU D Q. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age‐mediated flowering. Journal of integrative plant biology, 2020, 62(11): 1659-1673.
[21] Li W, Wang H, Yu D.WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under short-day conditions. Molecular plant, 2016, 9(11): 1492-1503.
[22] Kumar S V, Lucyshyn D, Jaeger K E, Alós e, Alvey e, Harberd n p, Wiggem p a. Transcription factor PIF4 controls the chemosensory activation of flowering. Nature, 2012, 484(7393): 242-245.
[23] CELESNIK H, ALI G S, ROBISON F M, REDDY A S N.VOZ (Vascular plant One-Zinc finger) transcription factors are required for proper regulation of flowering time. Biology open, 2013, 2(4): 424-431.
[24] PENG L T, SHI Z Y, LI L, SHENC G Z, ZHANG J L. Overexpression of transcription factor OsLFL1 delays flowering time in. Journal of plant physiology, 2008, 165(8): 876-885.
[25] ALTER P, BIRCHENEDER S, ZHOU L Z, SCHLÜTER U, GAHRTZ M, SONNEWALD U, DRESSELHAUS T. Flowering time-regulated genes in maize include the transcription factor ZmMADS1. Plant physiology, 2016, 172(1): 389-404.
[26] WENG L, BAI X D, ZHAO F F, LI R, XIAO H. Manipulation of flowering time and branching by overexpression of the tomato transcription factor Sl ZFP 2. Plant biotechnology journal, 2016, 14(12): 2310-2321.
[27] ASLAM M, JAKADA B H, FAKHER B, GREAVES J G, NIU X P, SU Z X, CHENG Y, CAO SJ, WANG X M, QIN Y. Genome-wide study of pineapple (L.) bHLH transcription factors indicates that cryptochrome-interacting bHLH2 (Ac CIB2) participates in flowering time regulation and abiotic stress response. BMC genomics, 2020, 21(1): 1-13.
[28] O’MALLEY R C, HUANG S C, SONG L, LEWSEY M G, BARTLETT A, NERY J R, GALLI M, GALLAVOTTI A, ECKER G R. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 2016, 165(5): 1280-1292.
[29] BARTLETT A, O'MALLEY R C, HUANG S C, GALLI M, NERY J R, GALLAVOTTI A, ECKER J R. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nature protocols, 2017, 12(8): 1659.
[30] STIGLIANI A, MARTIN-AREVALILLO R, LUCAS J, BESSY A, VINOS-POYO T, MIRONOVA V, VERNOUX T, DUMAS R, PARCY F. Capturing auxin response factors syntax using DNA binding models. Molecular plant, 2019, 12(6): 822-832.
[31] Galli M, Khakhar A, Lu Z, Sen S, Joshi T, Nemhauser J L, Schmitz R J, Gallavotti A. The DNA binding landscape of the maize AUXIN RESPONSE FACTOR family. Nature communications, 2018, 9(1): 1-14.
[32] LIANG S, GAO X X, WANG Y J, ZHANG H L, YIN K X, CHEN S L, ZHANG M, ZHAO R. Phytochrome-interacting factors regulate seedling growth through ABA signaling. Biochemical and biophysical research communications, 2020, 526(4): 1100-1105.
[33] 丁冬, 马拴红, 林源, 邱小倩, 万炯, 孟淑君, 王琪月, 张雪海, 汤继华. 玉米转录因子候选基因关联分析. 分子植物育种, 2021, 19(13):4206-4215.
DING D, MA S H, LI Y, QIU X Q, WAN J, MENG S J, WANG Q Y, ZHANG X H, TANG J H. Candidate genes association analysis of transcription factors in maize. Molecular Plant Breeding, 2021, 19(13): 4206-4215. (in Chinese)
[34] O’ MALLEY R C, HUANG S-S C, SONG L, LEWSEY M G, BARTLETT A, NERY J R, GALLI M, GALLAVOTTI A, ECKER J R, Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 2016. 165(5): 1280-1292.
[35] YANG N, LIU J, GAO Q, GUI S T, CHEN L, YANG L F, HUANG J, DENG T Q, LUO J Y, HE L J, WANG Y B, XU P W, PENG Y, SHI Z X, LAN L, MA Z Y, YANG X, ZHANG Q Q, BAI M Z, LI S, LI W Q, LIU L, JACKSON D, YAN J B. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nature genetics, 2019, 51(6): 1052-1059.
[36] XIAO Y J, TONG H, YANG X H, XU S Z, PAN Q C, QIAO F, RAIHAN M S, LUO Y, LIU H J, ZHANG X H, YANG N, WANG X Q, DENG M, JIN M L, ZHAO L J, LUO X, ZHOU Y, LI X, LIU J, ZHAN W, LIU N N, WANG H, CHEN G S, CAI Y, XU G, WANG W D, ZHENG D B, YAN J B. Genome‐wide dissection of the maize ear genetic architecture using multiple populations. New Phytologist, 2016, 210(3): 1095-1106.
[37] YANG N, LU Y L, YANG X H, HUANG J, ZHOU Y, ALI F, WEN W W, LIU J, LI J S, YAN J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genetics, 2014, 10(9): e1004573.
[38] HENDELMAN A, ZEBELL S, RODRIGUEZ-LEAL D, DUKLER N, ROBITAILLE G, WU X L, KOSTYUN J, TAL L, WANG P P, BARTLETT M E, ESHED Y, EFRONI I, LIPPMAN Z B. Conserved pleiotropy of an ancient planthomeobox gene uncovered by cis-regulatory dissection. Cell, 2021, 184(7): 1724-1739.
[39] NOH Y S, AMASINO R M. PIE1, an ISWI family gene, is required for FLC activation and floral repression in. The Plant Cell, 2003, 15(7): 1671-1682.
[40] BAI S, SUNG Z R. The role of EMF1 in regulating the vegetative and reproductive transition in(Brassicaceae). American journal of botany, 1995, 82(9): 1095-1103.
[41] YAN D W, ZHANG X M, ZHANG L, YE S H, ZENG L J, LIU J Y, LI Q, HE Z H. CURVED CHIMERIC PALEA 1 encoding an EMF 1‐like protein maintains epigenetic repression of O s MADS 58 in rice palea development. The Plant Journal, 2015, 82(1): 12-24.
[42] KIM W Y, HICKS K A, SOMERS D E. Independent roles for EARLY FLOWERING 3 and ZEITLUPE in the control of circadian timing, hypocotyl length, and flowering time. Plant physiology, 2005, 139(3): 1557-1569.
[43] DIXON L E, KNOX K, KOZMA-BOGNAR L, SOUTHERN M M, POKHILKO A, MILLAR A J. Temporal repression of core circadian genes is mediated through EARLY FLOWERING 3 in. Current Biology, 2011, 21(2): 120-125.
[44] KIM Y, YEOM M, KIM H, LIM J, KOO H J, HWANG D, SOMERS D, NAM H J. GIGANTEA and EARLY FLOWERING 4 inexhibit differential phase-specific genetic influences over a diurnal cycle. Molecular plant, 2012, 5(3): 678-687.
[45] ZHAO J M, HUANG X, OUYANG X H, CHEN W L, DU A P, ZHU L, WANG S G, DENG X W, LI S G. OsELF3-1, an ortholog ofearly flowering 3, regulates rice circadian rhythm and photoperiodic flowering. PLoS One, 2012, 7(8): e43705.
[46] ADEYEMO O S, KOLMOS E, TOHME J, CHAVARIAGA P, FREGENE M, DAVIS S J. Identification and characterization of the cassava core-clock gene EARLY FLOWERING 4. Tropical Plant Biology, 2011, 4(2): 117-125.
[47] Huang H, Gehan M A, Huss S E, Alvarez S, Lizarraga C, Gruebbling E L, Gierer J, Naldrett M J, Bindbeutel R K, Evans B S, Mockler T C, Nusinow D A. Cross-species complementation reveals conserved functions for EARLY FLOWERING 3 between monocots and dicots. Plant direct, 2017, 1(4): e00018.
[48] XIE Q G, WANG P, LIU X, YUAN L, WANG L B, ZHANG C G, LI Y L, XING H Y, ZHI L Y, YUE Z L, ZHAO C S, MCCLUNG C R, XU X D. LNK1 and LNK2 are transcriptional coactivators in thecircadian oscillator. The Plant Cell, 2014, 26(7): 2843-2857.
[49] 张庆雯, 祁静静, 谢宇, 谢竹, 彭蕴, 李强, 彭爱红, 邹修平, 何永睿, 陈善春, 姚利晓. 黄龙病菌胁迫下‘锦橙’CsCalS 表达和胼胝质沉积的初步分析. 园艺学报, 2021, 48(2): 276-288.
ZHANG Q W, QI J J, XIE Y, XIE Z, PENG Y, LI Q, PENG A H, ZOU X P, HE Y R, CHEN S C, YAO L X. Preliminary analysis of CsCalS5 and callose deposition in citrus sinensis infected with candidatus liberibacter asiaticus. Acta Horticulturae Sinica, 2021, 48(2): 276-288. (in Chinese)
[50] 崔海芳, 张凡, 尹俊龙, 郭瑛琪, 岳艳玲. 胼胝质沉积与花粉发育. 云南农业大学学报: 自然科学版, 2017, 32(3): 551-557.
CUI H F, ZHANG F, YIN J L, GUO Y Q, YUE Y L. Callose deposition and pollen development. Journal of Yunnan Agricultural University, 2017, 32(3): 551-557. (in Chinese)
[51] 杨俊. 拟南芥生长素响应因子ARF17调控花粉壁模式形成[D]. 上海: 上海师范大学, 2013.
YANG J.auxin response factor ARF17 regulates pollen wall pattern formation[D]. Shanghai: Shanghai Normal University, 2013. (in Chinese)
附表1 进行DAP-seq的转录因子信息
Supplementary Table 1 Transcription factor information for DAP seq
注:“附表2 22个玉米转录因子结合下游基因”因容量过大,不再赘列。如确有需要,请直接与作者联系。谢谢。
Candidate Gene Association Analysis of Maize Transcription Factors in flowering Time
MA ShuanHong1, WAN Jiong1, LIANG RuiQing2, ZHANG XueHai1, QIU XiaoQian1, MENG ShuJun1, XU NingKun1, LIN Yuan1, DANG KunTai1, WANG QiYue1, ZHAO JiaWen1, DING Dong1, TANG JiHua1
1College of Agronomy, Henan Agricultural University/National Key Laboratory of Wheat and Maize Crop Science, Zhengzhou 450002;2College of Agronomy, South China Agricultural University, Guangzhou 510642
【】Maize growth period traits, including flowering time, are the ones of most important in maize breeding. The advancement of heading date, silking time, and the pollen shed can ensure maize kernels fully dehydrated and thus suited to machinery harvesting. Moreover, the saved time can also leave for wheat sowing under the Maize-Wheat farming mode in Huang-Huai-Hai area. Transcription factors are important up-stream trans-action factors of gene expression regulation, which play roles in transcriptional activation or inhibition on target genes by binding to and driving their promoters. It is of great significance to analyze the regulatory effects of transcription factors on maize flowering time at the whole genome scale, it is also emergence to obtain the maize transcription factor haplotypes which associated with earlier flowering and higher yield. The haplotypes, or the haplotype combinations, will be served as excellent germplasm resources for maize breeding. 【】In this study, candidate gene association analysis was performed to analyze maize flowering time related transcription factors and significant SNPs. DAP-seq was carried out to obtain the binding sites and down-stream genes of the key transcription factors. Followed by GO analysis on the down-stream genes to explore the transcription factor dependent gene expression regulatory network. 【】There are 75, 75, and 128 significant SNPs detected in combinations of the traits Silking time and Heading date, the traits Silking time and Pollen shed, and the traits Heading date and Pollen shed, respectively. Altogether, there are 58 significant SNPs associated with all three flowering time traits. These results suggest that the three traits of flowering time may be regulated by the same transcription factors. Flowering time associated transcription factor genes that containing 3 or more significant SNPs were selected for DAP-seq to capture the key motifs and down-stream genes. Down-stream genes bound by flowering time associated transcription factors are significantly enriched in transcription factor activity, DNA binding, RNA binding, organonitrogen compound metabolic process, reproduction-related developmental processes, etc. Different transcription factors have co-regulated downstream genes related to flowering time. The key regulatory transcription factors for flowering time traits are ARF, MYB and NAC. Through haplotype analysis, the optimal TF haplotype combination that shows earlier flowering and no negative impact on yield was selected. 【】In this research, through candidate gene association and DAP-seq, the regulatory network of transcription factors on the flowering time related agronomic traits were established at the whole genome scale. The optimal haplotype combination of transcription factors that not only advances the flowering time, but also has no negative impact on yield was selected for further use in maize breeding.
maize; transcription factor; flowering time; DAP-seq; haplotype

10.3864/j.issn.0578-1752.2022.01.002
2021-07-07;
2021-09-03
国家自然科学基金面上项目(31871641,31971961)、作物遗传与种质创新国家重点实验室开放课题(ZW202001)、河南省科技攻关项目(202102110164,202102110012)
马拴红,E-mail:18838916904@163.com。通信作者丁冬,Tel:0371-56990336;E-mail:dingdong0216@hotmail.com。通信作者汤继华,Tel:0371-56990336;E-mail:tangjihua1@163.com
(责任编辑 李莉)
