掺杂镁、锌的铝团簇结构、稳定性和生长机制研究
2022-02-21韩雪薇
韩雪薇

摘要:铝合金具有高经济效益、高强度、低密度、再循环能力强等特点,被广泛用作结构材料。然而,铝合金具有应力腐蚀开裂缺陷。本文采用自旋极化密度泛函理论,对AlnM(M=Zn,Mg)(n=1-9)的结构、稳定性和生长机制进行研究。BLYP研究结果表明,M原子趋向于聚集在Al的表面上。除了Al7Zn和Al9Zn团簇,其同分异构体都倾向于密堆积特征。AlnZn的平均结合能最低。通过总结能差分析,可以发现Al3M (M=Al,Zn,Mg)和Al6M(Al,Zn,Mg)团簇结构更稳定。对AlnM(M=Zn,Mg)团簇,最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)能带隙的峰值在n=4,6,8,和n=6,9。AlnZn(n=1-4)的反应能最低。
关键词:铝团簇;掺杂;稳定性;生长机制
一、引言
铝合金这种有色金属结构材料,之所以在工业中应用相当广泛,是因其具有优良的特性。比如价格低廉、可回收、强度高、加工性能良好等。不仅在航空、航天、船舶等领域,在汽车、机械制造及化学工业中也被大量应用,是目前应用最多的合金。随着工业经济飞速发展,铝合金被应用于各行各业,对铝合金进行深入研究势在必行。Al-Zn-Mg-Cu合金是7000系列铝合金中一种超硬铝合金,可热处理,有良好的耐磨性和焊接性,但其应力腐蚀开裂缺陷限制了它的应用。众所周知,热加工工艺不同,热处理条件不同,Al-Zn-Mg-Cu铝合金的相不同、微观结构和性能等也会有变化。通过对锌、铜、镁元素进行显微分析,我们会发现短裂纹扩展路径偏折,这是因其合金中相邻晶粒的取向差较大导致其疲劳裂纹扩展,抗力减低,进而导致其性能较差。人们通常认为,影响硬化合金疲劳裂纹扩展的主要原因是微观结构,这就需要人们在实践中,控制化学成分的变化及其微观结构。我们通过研究锌、镁含量对7000系列铝合金微观结构和力学性能的影响可知,大多数高强度铝合金中,都存在细小、分散的不溶性金属化合物,如Zr、Mn、Cr、Ag、Ti、B等。所以实现晶粒细化可以进一步获得许多重要的性质[1]。这就是我们研究铝团簇掺杂镁、锌的结构、稳定性和生长机制的原因。
关于团簇的物理和化学性质,文献报道了铝团簇的几何结构、电子结构、电离势、电子亲和力、化学吸附性、光电子能谱等[5]。Rao和Jena采用BPW91 / lanl2dz方法全面研究了中性粒子、阳离子和阴离子铝团簇的平衡几何结构、电子结构。琼斯利用密度泛函计算与模拟退火方法计算了铝团簇(n = 10)的束缚能,并与实验结果进行比较。Li等人采用动态淬火模拟和多体势能分析研究了较大的铝中性团簇的几何结构和能量特性。获得全局最低能结构以及能量最低值的分布、每个粒子的有限温度热力学信息。Lu等人通过使用遗传算法方法探讨了原子数低于40个原子的铝原子的最低能结构,发现当团簇原子约为25-27个时出现有序分层结构。许多类型的元素已经被试作为替代品,如非金属(H、B、C、P、N等),活性金属(Na)和过渡金属(Ti、Fe、Ni、Cr、Mn等),Reuse等人计算了中性、阴离子、阳离子和双电离的镁团簇电子结构,研究电荷对几何结构和稳定性的影响。Kumar和Car等人采用密度泛函分子动力学方法和局域密度近似模拟退火技术研究了镁团簇的结构、生长机制、成键本质。也有人系统地计算了中性和单电荷镁团簇的几何结构、电子结构、结合能、电离势、电子最高占据轨道和最低未占有轨道间的能隙。更有人研究了中性粒子、阳离子和阴离子镁团簇的几何结构、稳定性的尺寸依赖关系和电学性质。但并没有关于系统研究铝掺杂镁团簇的报道,因此,进一步研究铝团簇掺杂镁是必要的。
二、计算方法
为了获得全局最低能结构,本项研究收集所有的数据库和出版物来找出所有可能的铝团簇的几何构型。确保我们没有跳过任何可能的结构。为了获得AlnM (M=Zn,Mg) (n=1-9)的初始结构,我们将Zn,Mg替换任何位置的铝原子。将所有假设的初始结构进行几何结构优化。我们的计算在DMol3模块中实施,采用自旋极化密度泛函理论(DFT),在广义梯度近似(GGA)下用BLYP函数进行计算,全电子密度泛函方法对团簇进行计算充分准确且非常有效[37]。选择双数值极化(DnP)基组。在几何结构优化过程中能量梯度和原子位移分别收敛于1×10 -5 Hartree/Bohr和 5×10 -3 Å。自洽迭代电荷密度收敛于1×10-6 e/Å3时,对应的总能量在1×10 -5 Hartree收敛。用布居分析分别计算获取的团簇原子电荷[38]。定义平均结合能为Eb(n)
其中:n是铝原子的数量。E(Al)和E(M)是铝团簇中单个原子的能量。E(AlnM)是铝团簇最低能量结构时的总能量。为了更好地理解铝团簇掺杂的相对稳定性,我们计算团簇能量的二阶差Δ2E。
其中:En+1 ,En-1 和 En分别代表Aln+1M, Aln-1M和 AlnM的结合能。为了解生长机制,只研究AlnM(n=1-9)能量最低的结构的AlmM+Aln-m → AlnM,定义为反应能量ΔE
三、结果与讨论
1. AlnM团簇结构
这是Aln(n=2-10)和AlnM(n=1-9)团簇最低能量结构,图中显示了Aln(n=2-10)团簇和AlnM(n=1-9)团簇的最低能量结构。从中可看出,AlnM团簇的结构受主体Aln(n=2-10)團簇结构的限制。随着团簇尺寸n的增大团簇发生了变化。M原子聚集在铝团簇的表面上倾向于高致密的堆积特征。特别是Al7Zn团簇,锌原子几何结构被优化到铝团簇的顶部位置。Al9Zn团簇中锌原子转换成与铝原子相邻的双原子。它证实了Al7Zn和Al9Zn团簇可以降低Al-Zn-Mg-Cu合金的力学性能。
Al2二聚体的键长是2.765Å,该计算值比实验结果略长,而比Rao和Jena的计算结果2.86 Å稍短。AlMg的键长为2.952 Å。而AlZn的键长则比AlMg的键长长。Al3团簇的基态结构为等边三角形,其键长是2.537Å,略短于Rao和Jena[15]的计算结果2.61 Å,且与Ren和Li的计算结果一致。Al2Mg和Al2Zn的结构是对称式C1等腰三角形。Al4团簇的基态结构是C1菱形结构对称的,其键长是2.657Å,该值略短于Rao和Jena的计算结果2.72Å。Al3Mg和Al3Zn的基态结构也均为菱形结构。掺杂锌、镁原子导致铝团簇的键长增加。而且掺杂锌、镁原子导致铝团簇结构发生扭曲,并使铝团簇从平面结构向四面体结构转变。从Al7Zn和Al9Zn可见,锌与铝团簇间的吸附能力较弱。
2. AlnM团簇的电子性质
团簇的稳定性可以用他们每个原子的平均结合能Eb和HOMO-LUMO(电子最高占有轨道-最低未占有轨道)之间的能隙来评估。我们通过公式来计算铝团簇的结合能。
合金的所有物理性质都与其电子结构息息相关。通过对铝团簇掺杂镁、锌HOMO态,M原子的电荷、原子自旋密度(电子数)计算分析表明AlnM团簇对电子的吸附能力依次为Al>Zn(0.034e)>Mg(0.114e)。Ouyang等人[39]证实了Mg原子的3s态和Al原子的3p态。
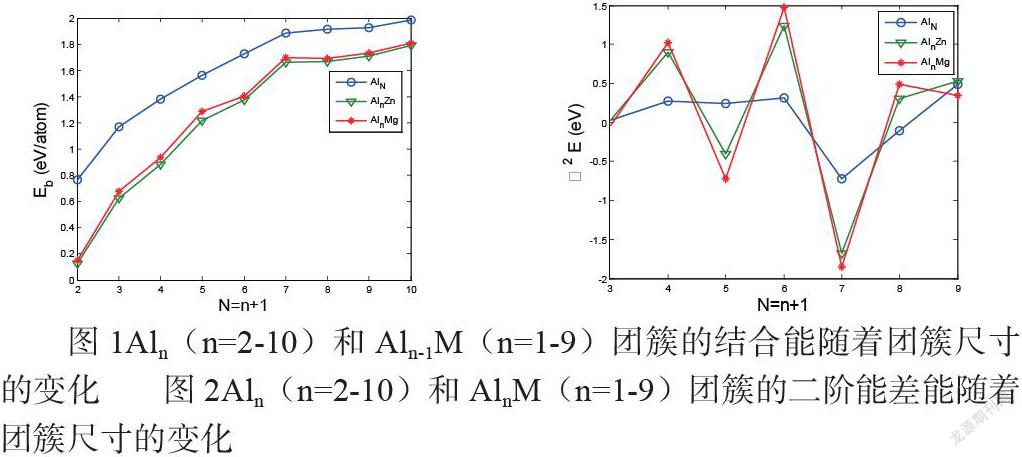
图1Aln(n=2-10)和 Aln-1M(n=1-9)团簇的结合能随着团簇尺寸的变化 图2Aln(n=2-10)和 AlnM(n=1-9)团簇的二阶能差能随着团簇尺寸的变化
图1显示为Aln(n=2-10)和Aln-1M (n=1-9)的结合能与原子数的关系。从中可看出,结合能随原子数的增加而增加。AlnM结合能的次序为Aln>AlnMg>AlnZn。Ouyang等人[39]已经证实了Aln-1Mg团簇比相应的纯铝团簇的原子结合能低。图2显示的是Aln (n=2-10) AlnM (n=1-9)和能量的二阶差。从中可看到,掺杂M(Zn, Mg)明显影响了铝团簇的相对稳定性。n=3,6时无论是纯铝团簇还是掺杂团簇其能量二阶差分值为正值,说明在我们所研究的团簇中,3个和6个原子的团簇相对于其他原子数的团簇更为稳定一些。[44]比较Aln (n=2-10) AlnM (n=1-9)团簇的能量二阶差曲线可以发现Al3M (M=Al,Zn,Mg)和Al6M (Al, Zn,Mg)团簇的结构比其它 AlnM (M=Al,Zn,Mg)团簇稳定得多。
能隙值是指电子最高占有轨道和最低未占有轨道之差,评价团簇的化学稳定性的一个重要指标即是能隙值。能隙值的大小可以直接反映团簇结构的稳定性,能隙值越大说明团簇越稳定。[5]图3
图3Aln(n=2-10)和 AlnM (n=1-9)团簇的HOMO-LUMO能隙随着团簇尺寸的变化 图4 Aln(n = 2 - 10)和AlnM (n=1-9)团簇的反应能随着团簇尺寸的变化
绘制了关于AlnM (n=1-9)和Aln(n=2-10)能隙大小的HOMO-LUMO曲线。从图3可看到,AlnM (M=Zn,Mg)团簇与纯铝团簇的HOMO-LUMO能隙曲線是不同的。对于Aln团簇,局部峰在n=1,3,8。对于AlnM (M=Zn, Mg)团簇,局部峰n= 4,6,8; n = 6,9。上述结论表明铝团簇掺杂镁、锌后其化学反应性优于纯铝团簇。
3. AlnM团簇的生长机制
为了了解AlnM团簇在较低的温度下的生长机制,我们观察了115优化组合。
从实验看出,小的Aln 团簇几乎都是Al原子在较低的温度下吸附得到的。然而,Al3M (Zn,Mg)与Al2-Al5团簇优先反应。Al5M (Zn,Mg)和Al4团簇优先反应。AlM (Zn,Mg)和Al2M (Zn,Mg)优先反应。团簇中n大小相同时,取最大的ΔE作为反应能。从图4可以看出,AlnZn (n=1-4)反应能是最低的,这意味着Al和Zn之间反应较弱。
四、结论
总之,通过用全电子密度泛函方法中,BLYP函数对AlnM (n=1-9)团簇进行空间结构、稳定性与生长机制的计算,结果表明,对于AlnM团簇最有利的结构是M原子趋向于聚集在铝团簇表面,除了Al7Zn和Al9Zn团簇,其同分异构体都倾向于高致密的堆积特征。AlnM结合Aln >AlnMg>AlnZn。通过分析,可以发现,Al3M (M=Al,Zn,Mg)和Al6M(Al,Zn,Mg)团簇结构更稳定。对于AlnM(M=Zn,Mg)团簇,最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)能带隙的峰值在n=4,6,8,和n=6,9。AlnZn(n=1-4)的反应能最低。
参考文献:
[1]Y. L. Wu, C. G. Li, F. H. Froes, A. Alvarez. Microalloying of Sc, ni, and Ce in an advanced Al-Zn-Mg-Cu alloy. Metall Mater Trans A. 1999,30,1017.
[2] G.W. Turner, R.L. Johnston, n.T.Wilson. Investigation of geometric shell aluminum clusters using the Gupta many-body potential. J. Chem. Phys.2000,112,4773–4778.
[3] J. Akola, M. Manninen, H. Häkkinen, U. Landman, X. Li, L.S. Wang. Aluminum clusters anions: photoelectron spectroscopy and ab initio simulations. Phys. Rev. B. 2000,62,13216–13228.
[4]王广厚. 原子团簇的稳定结构和幻数. 物理学进展.2000,20,52
[5]张微.铝团簇的结构、稳定性和电子性质的理论研究.吉林大学.2009,55
