MOCVD生长InN气相反应路径的量子化学研究
2022-02-18何晓崐薛园左然
何晓崐,薛园,左然
(1 江苏科技大学苏州理工学院,江苏 张家港 215600; 2 密西西比大学化学与生物化学系,密西西比 牛津 38677;3欧柏林学院化学与生物化学系,俄亥俄 欧柏林 44074; 4 江苏大学能源与动力工程学院,江苏 镇江 212013)
引 言
Ⅲ族氮化物半导体材料(包括GaN、InN、AlN 及其二元和三元合金)是制备光电子器件和电力电子器件的关键材料,金属有机化学气相沉积(MOCVD)是生长Ⅲ族氮化物半导体薄膜的主要技术[1-2]。在GaN、InN、AlN 三种氮化物中,InN 具有最低的化学键能,造成InN 的热解温度低(约630℃),N 平衡压强高。在MOCVD生长InN薄膜时,必须采取远低于GaN 和AlN 生长的温度以及N2作为载气。为了使NH3在较低温下能够分解出足够的N 原子,又必须采用极高的Ⅴ/Ⅲ比(通常大于10000)来促进NH3的分解[3]。因此,尽管高In 组分对于制备LED 和激光器的量子阱结构以及微波器件和电力电子器件的异质结至关重要,然而具有高均匀性和高In 组分的InN和InGaN薄膜生长目前仍很困难。
关于Ⅲ族氮化物MOCVD 生长的化学反应路径,国内外已经进行了大量的研究,但大多数都围绕GaN 和AlN 的MOCVD 生长[4-8],关于InN 生长的化学反应路径仍了解不多。在InN的MOCVD生长中,In(CH3)3(简称TMIn)与NH3在载气携带下进入反应腔,在高温激活下,反应气体发生气相和表面反应,在衬底表面形成InN 薄膜。对于MOCVD 生长InN的气相反应路径,目前主要有两种观点。第一种观点认为,TMIn 在高温下通过热解逐步消去CH3,变成InCH3(简称MMIn)或In,即热解路径占主导。Creighton 等[9]利用傅里叶变换红外光谱分析(FTIR)发现,TMIn与NH3相遇后首先生成加合物TMIn:NH3,但随着温度升高,TMIn:NH3重新可逆分解为TMIn与NH3,TMIn 进一步热解为In(CH3)2(简称DMIn)、InCH3和In。Sekiguchi 等[10]对TMIn 在H2气氛下的反应进行了量子化学计算,通过对比各反应的ΔG,发现绝大部分TMIn 通过热解反应生成气相In 原子,少部分生成MMIn。第二种观点认为,TMIn 与NH3生成加合物TMIn:NH3后,不可逆分解为氨基物DMInNH2及其后续产物,即加合路径占主导。Nakamura 等[11]利用量子化学计算,发现加合物TMIn:NH3会通过单分子反应以及与TMIn 发生分子间反应消去CH4,生成氨基物DMInNH2,从而削弱了TMIn:NH3可逆分解的反应。上述结果与Thon 等[12]利用原位光谱技术对TMGa 与NH3反应的研究结果相似。
上述InN 生长的两条气相反应路径均未考虑气相反应中的自由基作用。在Ⅲ族氮化物的MOCVD生长中,存在着CH3、H和NH2自由基,特别是H自由基的活性很强,将积极地参与反应,造成反应路径的变化。Cavallotti 等[13]对MOCVD 生长GaN 的气相反应进行量子化学计算研究,认为当H2作载气时,H2很容易与TMGa 热解产生的CH3反应,生成H 自由基,而H 自由基又会加速TMGa 的热解。McDaniel 等[14]利用实验并结合数值计算研究了TMIn 分别在He、H2、D2中的热解过程,发现当载气全部为He时,TMIn热解后产生的只有C2H6,无CH4,显然此时的C2H6来自TMIn 热解出来的两个CH3。但当载气中混入H2或D2之后,则有CH4或CH3D 产生,即H2提供了CH4中的H,证明H2参与并加速了TMIn 的热解。Creighton 等[15]通过对GaN 生长的衬底前沿的纳米粒子薄层进行激光散射和质谱测量,发现该薄层对H2作载气时散射强度大,对N2作载气时散射强度迅速下降。由于散射强度对应纳米粒子浓度,因此证明H 自由基对热解反应和加合反应都起到了加速作用。Zhang等[16]通过CFD模拟证明,自由基主要产生在高温衬底附近,NH2自由基会与TMGa 迅速反应生成DMGaNH2,因此GaN 气相反应为自由基参与的加合路径主导。Moscatelli 等[17]利用量子化学计算,研究了NH2自由基在MOCVD 生长GaN 中的作用,发现NH2自由基会与TMGa 反应生成DMGaNH2等氨基物,而DMGaNH2会发生聚合反应,最终形成纳米粒子,因此DMGaNH2被认为是纳米粒子形成的基元,由此得出自由基会加剧GaN生长过程中的氨基反应。
虽然国内外学者对MOCVD 生长Ⅲ族氮化物进行过大量理论和实验研究[18-19],但对MOCVD 生长InN 的气相反应仍了解不多,尤其是不同温度下自由基对InN 生长的气相反应路径的影响研究很少。本文利用量子化学计算,对自由基作用下MOCVD生长InN 过程中TMIn 的热解反应路径、TMIn 与NH3的加合反应路径进行计算,通过对比不同温度下各反应的Gibbs 能差ΔG和反应能垒ΔG*/RT,分别从热力学和动力学上探索MOCVD 生长InN 的气相反应机理。
1 计算模型及验证
结合前人的研究结果[9,11,14,17],本文提出在富NH3条件下,MOCVD 生长InN 的气相反应机理,如图1所示。由于NH3在MOCVD 温度下不能直接热解,因此只考虑从TMIn 开始的反应路径,包括TMIn、DMIn 等与自由基H、NH2的反应。产生自由基的反应如文献[13]给出,本文假设上述自由基存在于H2载气中。除自由基参与的热解、加合路径外,还包括氨基物DMInNH2的聚合路径,即DMInNH2之间发生二聚和三聚反应,最终生成纳米粒子[15,20-21]。

图1 MOCVD生长InN的气相反应路径示意图— 热解路径; 加合路径; H自由基参与路径; NH2自由基参与路径; 聚合路径Fig.1 Schematic of InN MOCVD gas-phase reaction pathway
本文采用Gaussian 09[22]进行计算,TMIn 与NH3反应生成InN 过程中涉及的反应物、过渡态、中间体、产物等的结构优化、频率计算采用密度泛函理论(DFT)中的M062X 方法,该方法比B3LYP 方法更适合计算有机体系,计算基组为6-31G(d)。金属In原子的内层电子采用赝势,外层电子采用LANL2 DZ。为了确保得到的过渡态连接反应物和产物,通过内禀反应坐标(IRC)进行验证。在获得优化结构后,再利用M062X/6-311G(d,p)对所有物质进行单点能计算,以获得更高精度的电子能。在计算得到的单点能和优化结构基础上,利用Shermo 程序,计算不同温度下的热力学数据,包括:内能E、焓H、熵S、Gibbs 能G等。Shermo 是 由Lu 等[23]开 发 的 计 算 分 子热力学数据的开源程序。考虑到Gaussian计算假设分子作刚性谐振动,而实际分子存在非谐振动,因此在使用Shermo 程序时,引入频率校正因子。根据Truhlar等[24]的拟合结果,采用0.967作为本文计算的零点能(ZPE)校正因子。焓和熵的校正因子都接近1,故直接设定为1。优化后的主要物质的分子结构如图2所示。

图2 优化后的InN MOCVD过程中的主要气相物质的分子结构Fig.2 Optimized molecular structures of major gas species in InN MOCVD process
在MOCVD 反应腔中,从室温进口到高温衬底,气体温度变化很大。因此,气相反应也随气体在反应腔的位置而改变。由于反应腔中压力变化很小,因此反应路径主要依赖于腔内的局域温度。计算中,设定压强为1 atm(1 atm=101.325 kPa),该压强接近真实的MOCVD 过程(0.1~1 atm)。假设气体充分混合,并且气体的驻留时间足以保证化学反应的完成。
1 mol理想气体的内能E、焓H、熵S和Gibbs能G的关系式如下:

其中,Ee、Etran、Erot、Evir分别为分子处于基态时的电子能、平动能、转动能、振动能;ZPE为分子的零点能;E0为0 K时分子的内能。
为了预测化学反应的发生和快慢,本文将不同温度下各反应物总的Gibbs 能初始值均设为0,通过产物与反应物的Gibbs 能差ΔG的正负,来判断反应进行的方向(热力学判据)。通过反应能垒ΔG*/RT的大小来预测反应速率[25](动力学判据)。其中ΔG*为过渡态相对反应物的Gibbs 能差,R为气体常数。反应能垒ΔG*/RT越小,则反应速率越大。用于预测反应路径的Gibbs能判据如图3所示。

图3 反应过程的Gibbs能变化和判据Fig.3 Schematic diagrams of the Gibbs energy changes and the criteria of determining the reaction process
由于加合物TMIn:NH3和氨基物DMInNH2在InN 生长的气相反应中被广泛研究,因此选择这两种物质的计算结果与文献值进行对比,以验证计算结果的准确性,结果列于表1。同时,本文还将涉及该两种物质的反应G4、G6 的ΔH值与文献值进行了对比,结果见表2。由表1 和表2 可见,计算值与文献值对应良好,误差主要来自选用计算方法、基组的不同。

表1 优化后的TMIn:NH3、DMInNH2键长和键角对比Table 1 Comparisons of optimized bond lengths and bond angles for TMIn:NH3 and DMInNH2

表2 反应G4、G6的ΔH的计算值与文献值的对比Table 2 Comparisons of the ΔH of reactions G4, G6 calculated in this study and reported from literatures
2 结果与讨论
2.1 直接热解路径
当无自由基参与时,TMIn 直接热解,分步脱去CH3的反应为:

表3 所示为不同温度下TMIn 热解反应的Gibbs能差ΔG。反应G1~G3 均无过渡态,在所研究温度范围内,反应G1 的Gibbs 能差ΔG均为正值,在低温下ΔG很大,因此低温时TMIn热解很难进行;但随着温度升高,ΔG逐渐降低,当温度从T=298.15 K 升高到T=1073.15 K 时,ΔG从59.03降低至30.12,因此高温有利于反应G1 的发生。对反应G2,当T>873.15 K 时,ΔG<0,此时反应G2 可自发进行。反应G3 与G1 类似,在低温时很难热解,在高温时由于反应的ΔG下降,有利于热解的发生。由此得出以下结论:TMIn 和MMIn 需要在高温下才能热解,而DMIn 热解不需要很高的温度。

表3 TMIn热解反应的Gibbs能差ΔGTable 3 The changes of Gibbs energy ΔG in TMIn pyrolysis
为了对比TMIn 与TMGa 热解的难易程度,取计算得出的TMIn 热解反应的焓变ΔH与对应的TMGa热解反应数据[6]进行对比。TMIn 分步热解为DMIn、MMIn、In(反应G1~G3)的ΔH(298.15 K)分别为:69.5、27.7、55.4 kcal/mol。对应298.15 K 时TMGa 分步热解为DMGa、MMGa、Ga 的ΔH的量子化学计算数据为:74.4、34.1、62.6 kcal/mol。由此可见,TMIn比TMGa 更容易热解,这从In-C 键长(2.15 Å)大于Ga-C 键长(1.98 Å)、In-C 键能低于Ga-C 键能可以推出上述结论。
2.2 加合路径
大量研究[9,11,29]证明,TMIn 与NH3相遇后立刻生成Lewis酸碱加合物TMIn:NH3。TMIn:NH3可能发生可逆分解,重新生成TMIn 和NH3;也可能发生消去CH4的不可逆分解,生成氨基物DMInNH2。TMIn 也可直接与NH3反应生成DMInNH2。反应如下:
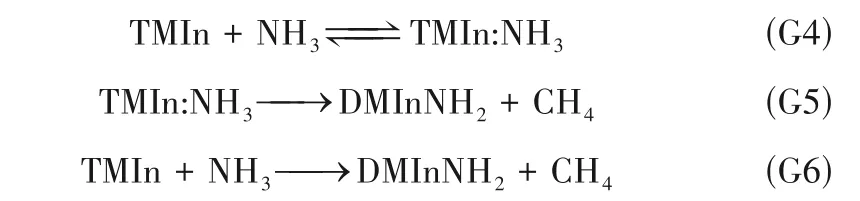
图4 所示为不同温度下,TMIn 与NH3加合反应路径的Gibbs 能垒变化。由图4 可见,当温度低于473.15 K 时,反应G4 的Gibbs 能差ΔG<0。通过插值计算,当T≈602.4 K 时,ΔG=0。因此,当T<602.4 K 时,反应G4 正向自发进行,当T>602.4 K 时,TMIn:NH3倾向于可逆分解为TMIn 和NH3。反应G5的Gibbs 能差ΔG<0,即反应能自发生成氨基物DMInNH2,但需越过能垒ΔG*/RT。反应能垒随温度升高明显降低。反应G6 的Gibbs 能差ΔG<0,其过渡态与反应G5 的过渡态重合,但不经过加合路径。因此TMIn 也可与NH3直接反应生成氨基物DMInNH2,但需要越过33.1~68.2(T=1073~300 K)的无量纲反应能垒,反应能垒随温度升高而降低,高温更有利于G6发生。显然,加合路径(G4~G6)与直接热解路径(G1~G3)存在竞争关系。与GaN生长相似,两种路径的强弱取决于反应室压强、气体的混合程度以及温度梯度的大小[30]。显然低压强、低混合程度,有利于热解路径,反之则有利于加合路径。温度梯度小,有利于TMIn:NH3可逆分解为TMIn 与NH3,TMIn 进一步加热则发生热解;温度梯度大,则有利于反应G5、G6 克服能垒,不可逆分解为DMInNH2,即有利于加合路径。

图4 加合路径的ΔG/RT变化(弧线代表反应G6)Fig.4 The changes of ΔG/RT in the adduct reaction path (the arcs represent reaction G6)
2.3 H自由基参与的反应路径
根据前人研究,当H2作为载气时,TMIn 热解产生的CH3自由基,会与H2反应生成H 自由基[13,31];即使载气全部为N2,NH3也会由于高温热解或表面反应生成H2。H 自由基将加速烷基铟(TMIn、DMIn、MMIn)的热解[13]。H 自由基参与的TMIn 热解反应如下:

图5 所示为H 自由基参与的热解反应的Gibbs能垒变化。在所研究温度范围内,反应G7~G9 的ΔG均小于0,表明反应均可自发进行;而TMIn 直接热解反应(G1~G3)则不能自发进行,说明自由基可明显促进TMIn 的热解。反应G7~G9 均存在过渡态,过渡态能垒ΔG*/RT均随温度的升高而降低,表明高温有利于反应进行。因此,在典型的InN 或InGaN MOCVD 生长温度下,由于H 自由基的参与,TMIn 在高温衬底附近会快速热解,最终生成气相In 原子,与前人对GaN 的研究结果一致[17]。

图5 TMIn及其热解产物与H自由基反应的ΔG/RT变化Fig.5 The changes of ΔG/RT in TMIn pyrolysis path with radical H
氨基物DMInNH2也可与H 自由基反应,逐步消去CH4,反应式如下:
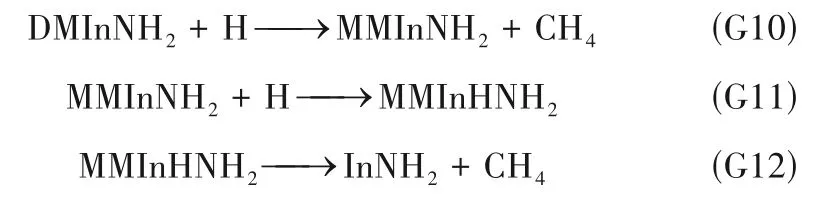
图6 为反应G10~G12 的反应能垒变化,在所研究温度范围内,反应G10~G12 的ΔG均小于0,反应均可自发进行。反应G10 和G12 的反应能垒ΔG*/RT随温度升高而降低,表明高温有利于反应进行。反 应G11 无 能 垒,MMInNH2与H 反 应 生 成 的MMInHNH2可视为反应中间体,该中间体可通过反应G12 分解为InNH2和CH4。G12 的无量纲反应能垒ΔG*/RT在低温(298 K)时很大,约为165;当温度升高至1073 K 时,ΔG*/RT减小至约30。因此,在自由基H 的作用下,InNH2能够在高温衬底附近产生,从而使表面反应前体由传统的MMIn 和In 变为InNH2。

图6 DMInNH2与H自由基反应的ΔG/RTFig.6 The changes of ΔG/RT in DMInNH2 reactions with radical H
2.4 NH2自由基参与的反应路径
根据前人研究得知,H 自由基很容易与NH3发生反应生成NH2自由基[16,31],NH2能够与TMIn及其热解产物DMIn、MMIn、In发生反应,反应式如下:

自由基反应G13~G18均无过渡态,由表4可见,G13~G18 的Gibbs 能差ΔG<0,因此所有反应均可自发进行。G13~G15 的Gibbs 能差ΔG随温度升高变化不大,但G16~G18 的ΔG的绝对值随温度升高而减小(即负值增大),表明随温度升高,反应概率降低。

表4 TMIn及其热解产物与NH2自由基反应的Gibbs能差ΔGTable 4 The changes of Gibbs energy ΔG in TMIn pyrolysis with NH2 radical
氨基物DMInNH2除了通过反应G13和G16产生外,也可通过反应G5和G6产生。若载气中无H2,此时DMInNH2主要通过反应G5和G6生成。若载气中有大量H2,热解产生的CH3与H2反应,产生H 自由基,H 自由基又与NH3反应生成NH2。此时NH2自由基的量相对较大,反应G13~G18 将占主导,由此产生各种氨基物DMInNH2、MMInNH2等,反应可以在离高温衬底较远区域发生。此时,DMInNH2很容易进一步聚合为多聚物,最终形成纳米粒子。两种反应均可在距离高温衬底较远处进行。由于H2和NH3与TMIn的浓度比通常极大,因此氨基物的聚合反应可能占据主导。由此可知,H2载气会加剧InN 生长的寄生反应,与前人研究结果[13,15]一致。这一结果也提供了InN/InGaN 生长中极少采用H2作为载气的一个原因。
2.5 氨基物聚合路径
大量研究表明,DMGaNH2和DMAlNH2会发生聚合反应最终生成纳米粒子[15,20-21],因此,DMInNH2可能会发生类似反应。常见的聚合反应为二聚反应和三聚反应:

表5 为不同温度下,反应G19 和G20 的Gibbs 能差ΔG。由表5 可见,在所研究温度范围内,G19 的ΔG<0,反应可自发进行。对于G20,ΔG在673.15 K和873.15 K 之间由负变正,利用线性插值计算,得T≈776.2 K 时,ΔG=0,即当T<776.2 K 时,反应G20能够自发进行。由于聚合反应无能垒,因此氨基物DMInNH2形成后会快速变为二聚物(DMInNH2)2,二聚物在T<776.2 K 时可形成三聚物(DMInNH2)3,最后形成纳米粒子。由于MOCVD 生长InN 的温度相对较低(约873.0 K),因此反应腔内有较大区域T<776.2 K,聚合反应很容易发生。根据前人研究[32],TMIn 在680.0 K 以上发生热解产生CH3。由2.4 节分析可知,当载气中有H2时,CH3将与H2反应,生成H 自由基;H 自由基继续与NH3反应,生成NH2自由基。即一旦有CH3产生,NH2也会快速生成,生成区域温度也为680.0 K 左右。NH2自由基很容易通过反应G13 和G16 形成DMInNH2,在此温度下,DMInNH2将快速聚合为(DMInNH2)3,继而形成纳米粒子。

表5 DMInNH2二聚反应(G19)和三聚反应(G20)的Gibbs能差ΔGTable 5 The changes of Gibbs energy ΔG in dimerization(G19) and trimerization (G20) of DMInNH2
综上,当载气中有H2时,MOCVD 生长InN 的气相反应路径以TMIn、DMIn 与NH2自由基的反应以及氨基物DMInNH2的聚合反应为主导;当载气中无H2(纯N2)时,DMInNH2主要通过加合路径(G4~G6)生成,由于反应G5 和G6 能垒较大,需要高温激活,而高温不利于聚合反应,因此DMInNH2不易聚合生成(DMInNH2)3,此时InN 生长的气相反应为热解路径与加合路径的相互竞争,具体路径取决于压强和温度梯度等反应器参数。
3 结 论
本文利用量子化学的密度泛函理论,对MOCVD 生长InN 的气相反应路径进行较全面的计算分析,特别是研究了自由基对InN 的气相反应路径的影响。结论如下。
(1)TMIn 直接热解反应的Gibbs 能差ΔG>0,反应不能自发进行。但在H 自由基的参与下,从TMIn到In 的热解反应均可自发进行,H 自由基促进了TMIn的热解反应。
(2)当载气为N2时,MOCVD 生长InN 的气相反应路径主要为热解路径与加合路径的竞争。当T<602.4 K 时,TMIn 与NH3反应生成加合物TMIn:NH3;当T>602.4 K 时,TMIn:NH3可能可逆分解为TMIn 和NH3,TMIn在高温将继续热解,也可能不可逆分解为氨基物DMInNH2和CH4。具体的反应路径取决于反应器压强和温度梯度。低压强、小温度梯度有利于热解路径;反之则有利于加合路径。
(3)当载气为H2时,H自由基与NH3反应形成大量NH2自由基。NH2又可与TMIn热解反应产物生成氨基物DMInNH2。因此H自由基和NH2自由基同时促进了TMIn 的热解反应和氨基物DMInNH2的生成反应。由于自由基反应不需要高温,因此两种反应均可在距离高温衬底较远处进行。
(4)DMInNH2可与H 自由基反应,在高温衬底附近生成InNH2,使表面反应前体由MMIn 和In 变为InNH2。在T<776.2 K 的区域,DMInNH2还会快速聚合为三聚物(DMInNH2)3,继而形成纳米粒子。因此H2会促进InNH2表面前体的产生,同时加剧纳米粒子形成。纳米粒子的产生不利于薄膜生长,但InNH2表面前体对薄膜生长的影响仍有待研究。
