色谱与MC-ICP-MS联用在线同位素分析的研究进展
2022-02-18杜媛媛朱振利郑洪涛
杜媛媛,朱振利*,郑洪涛,刘 星
(1.中国地质大学(武汉) 生物地质与环境地质国家重点实验室,湖北 武汉 430078;2.中国地质大学(武汉) 材料与化学学院,湖北 武汉 430078)
近年来,随着环境污染愈发严重,人们越来越关注各种金属和非金属元素对环境和人体健康的影响和危害。早期大多数研究只对元素的总量进行分析,但越来越多的研究表明只分析元素的总量难以真实反映元素的影响。这主要是因为元素在迁移过程中会发生形态的转化,而这些元素的赋存形态显著影响元素的毒性及其迁移转化过程。例如,汞的化学形态控制着其生物可利用度、迁移、持久性和对人体的影响,通常有机汞的毒性大于无机汞,其中甲基汞的毒性最大。因此,世界卫生组织制定的水生环境和海产品中甲基汞的浓度限制比无机汞(Hg2+)更严格[1]。此外,还有很多元素(如锑、铅、镉、卤素等)的不同形态具有不同的毒性和化学过程。因此,对样品中存在的某种元素的不同化学形态进行鉴别和测定,即形态分析是很有必要的。
另一方面,各种元素的同位素由于其自然或人为过程引起的丰度变化的特殊特征,已逐渐发展成为地球科学、环境和生命科学等领域中广泛应用的示踪剂[2]。使用同位素比值质谱(IRMS)测量传统稳定同位素(H、C、O、N、S)已成功应用了几十年,科学家通过研究它们在许多不同环境中的环境循环,解决了地质、环境研究中的诸多问题。随着热电离质谱(TIMS)与多接收电感耦合等离子体质谱(MC-ICP-MS)的迅速发展,非传统稳定同位素以及放射性成因的金属同位素的分析成为可能并日益取得了广泛应用[3-4]。例如,镉在非污染的自然环境系统和人为来源中的同位素组成一般具有不同的同位素特征,镉同位素作为一种新型的示踪剂,可用于研究古环境和追踪镉循环,为环境研究提供了一种有前景的源示踪技术[5-6]。汞稳定同位素的自然丰度测量已成为区分汞源和理解环境中汞的生物地球化学过程的强有力的示踪剂[7]。除了非传统稳定同位素,Pb、Sr、Nd、Hf 等放射性成因的金属同位素体系也被广泛应用于溯源与示踪研究。例如由于Pb同位素之间的质量差异相对较小,在地球表面条件下,Pb在物理和化学过程中不会发生较大的同位素分馏,因此铅同位素已被广泛用于环境中各种铅污染物的来源和途径确定[8]。与TIMS相比,MC-ICP-MS具有更高的电离效率和更低的检出限,可接受的基体效应和分馏效应等优点[9]。而且MC-ICP-MS的样品引入装置范围广泛,与必须“脱机”引入样品的TIMS 不同,MC-ICP-MS 允许连续引入样品到ICP 源。MC-ICP-MS 的这些特性使其逐渐成为非传统稳定同位素以及放射性成因同位素分析的主流方法。
利用MC-ICP-MS进行高精度同位素分析,通常需采用离子交换树脂将待测元素与干扰元素、基体元素有效分离后进行离线测定,非常耗时,大大影响了同位素样品的分析效率。色谱与MC-ICPMS联用可实现样品中基体和干扰元素的在线分离,显著减少了样品前处理时间,节省了成本,减小了样品分离纯化过程中可能产生的交叉污染,使得快速获取高精度同位素成为可能,因此日益引起研究者的关注。另一方面,元素形态转化过程中伴随着同位素分馏过程,研究表明稳定同位素的单体同位素可以提供更有效、更丰富的信息,逐渐成为研究热点[10]。进行形态同位素分析同样需要将元素的不同形态分离开,然后进行不同元素形态的同位素比值测定。形态分离通常采用色谱分离的方式,这种色谱与MC-ICP-MS联用进行形态同位素在线分析的技术近年来日益受到关注。通过分析不同元素的形态同位素,可以了解不同元素形态的生物地球化学循环过程,为了解和解决地质、生命和环境问题提供思路和工具。
目前已有许多研究报道开发了各种色谱与MC-ICP-MS联用技术,实现了不同元素及其形态同位素测定,主要包括气相色谱(GC)、液相色谱(LC)、毛细管电泳(CE)及离子色谱(IC)等(如图1)。Krupp 等[11-12]首次报道了将毛细管气相色谱(CGC)与MC-ICP-MS 联用(CGC/MC-ICP-MS)实现PbEt4铅同位素比值测定。通过一个T 型管引入铊内标溶液校正质量歧视效应,通过峰面积计算同位素比值,实现了对瞬态信号的精确测定,高丰度同位素比值的精度为0.02%~0.07%。此后的二十年,陆续有越来越多的研究发展了色谱与MC-ICP-MS 联用技术,实现了许多同位素体系的在线分离和检测,如卤素、汞、锑、铅、硫等。尽管色谱与MC-ICP-MS联用的应用仍然不是十分广泛,存在许多待解决的问题,但色谱与MC-ICP-MS联用已在元素形态同位素以及快速同位素分析方面取得了显著进展,引起了研究者的广泛兴趣。本文查阅近年来的相关文献,简要回顾了色谱与MC-ICP-MS联用测定形态同位素比值的历史,讨论了影响在线同位素分析的瞬态信号采集和处理策略以及质量歧视校正方法,并从分析特性、优缺点以及今后在环境、化学等研究中的应用等方面进行了概述。

图1 色谱与MC-ICP-MS联用Fig.1 Chromatography coupled to MC-ICP-MS
1 瞬态信号处理
金属同位素分析通常采用离线方法富集纯化样品,然后使用MC-ICP-MS 通过连续稳定进样来测定同位素比值,获得一个连续稳定的信号。然而,色谱与MC-ICP-MS 联用在线分离测定同位素比值通常获得的是瞬态信号,这对高精度同位素分析带来了挑战。处理瞬态信号的难点主要在于数据收集时间短,分析物瞬态流动过程中同位素比值的漂移以及数据处理的复杂性[13]。
计算瞬态信号中同位素比值的常用方法有峰面积积分法(PAI)、逐点计算法(PbP)和线性回归斜率法(LRS)。峰面积积分法又分为全峰积分法和峰中心积分法。色谱与MC-ICP-MS 联用时,瞬态信号的强度随时间的变化曲线(色谱峰)通常是高斯型曲线。全峰积分法是指在整个峰值持续时间内(基线到基线),使用一定的积分时间对瞬态信号值进行采样,积分不同同位素的信号,它们积分数据的比值即为同位素比值(如图2A)。峰中心积分法是指只对峰中心部分(一定峰宽内从左至右)进行积分,分别积分不同同位素的信号,它们积分数据的比值即为同位素比值(如图2B)。逐点计算法分为基于非加权和加权信号点求和的计算。假设元素E 有2 个同位素分别为xE和yE,使用非加权信号点求和时,逐点计算的公式[14]为:

图2 峰面积积分法和线性回归斜率法的示意图Fig.2 Schematic diagram of peak area integration and linear regression slope method

其中,x/yE为同位素比值;RtS和RtE分别为用于计算的保留时间开始和结束的点;n为用于计算的信号强度点数。即在保留时间内在每个信号测量点计算同位素比值以及比值总和,然后除以使用的测量点数量,得到同位素比值。与非加权信号点求和不同,加权信号点求和考虑了信号强度随时间的变化,通常会有更高的精度,使用加权信号点求和的逐点计算公式[14]为:

Van Acker等[15]使用GC/MC-ICP-MS测定含氯脂肪族碳氢化合物中的氯同位素比值时,采用全峰积分法处理瞬态信号,发现峰尾信号对同位素比值的准确性有重要影响。因此,采用全峰积分法时需要准确确定色谱峰的起始和结束位置,尤其是拖尾的色谱峰,保留色谱峰的整个峰尾直至背景上没有可识别的信号才能确保精度和准确度。Karasinski 等[16]采用IC/MC-ICP-MS 测定镁同位素比值时,通过IC将Mg与干扰基体分离,采用全峰积分法处理瞬态信号,得到了较好的精度(约为0.15‰,2SD)。
Wehmeier 等[17]通过GC/MC-ICP-MS 测定三甲基锑中锑的同位素比值时,采用峰面积积分法和逐点计算法分别处理瞬态信号,计算同位素比值。结果显示采用梯形面积积分法对全峰和峰中心积分可获得最佳精度,相对标准偏差(RSD)分别为0.08%和0.02%。其中,峰中心积分法的精度更好,这是因为采用全峰积分法,减去背景基线时会引入误差,而这种误差在小信号(如峰尾端)时尤为明显。但研究者也指出,采用峰中心积分法得到准确同位素比值需要色谱分馏是围绕峰中心对称分布的。
线性回归斜率法是指在处理瞬态信号时,对元素的两个同位素信号强度进行线性拟合,这些数据点的最佳线性拟合的斜率即表示同位素比值(如图2C,横坐标和纵坐标的yE 和xE 分别表示同位素yE和xE的信号强度)。
Queipo-Abad 等[18]用GC/MC-ICP-MS 测定汞形态同位素组成时,对峰面积积分法、逐点计算法和线性回归斜率法计算同位素比值进行了比较。3 种方法分别计算了在窄峰(2~5 s)和宽峰(20~25 s)条件下多个汞同位素的δ值。当使用窄GC 峰值时,测量以较短的积分时间执行(0.131 s 和0.262 s),而当使用较宽的峰值时,则使用较长的积分时间(0.524 s和1.049 s)。在不同的实验条件下,独立测量了δ202Hg(Ⅱ)的平均值(‰)(n=8)。结果表明,采用线性回归斜率法,在321~641 个采集点之间选择较窄的气相色谱峰,获得了最佳的外精度。在此条件下,所有δ值的2SD 范围为0.236‰~0.590‰。结果表明,使用线性回归斜率法时,从背景中包含足够数量的采集点至关重要。当使用较窄的峰时获得了更好的外部再现性,这可能是由于在处理窄峰时,峰值的灵敏度更高,积分时间更短,数据点数量增加。
Horst等[19]采用GC/MC-ICP-MS 测定挥发性脂肪族化合物中δ37Cl,分别采用线性回归斜率法和常用的面积积分法计算同位素比值。采用线性回归斜率法时,采集在瞬态信号的每个时间点上37Cl和35Cl的信号强度并进行线性拟合,最佳线性拟合的斜率表示同位素比值37/35Cl;采用面积积分法时,37/35Cl比值的计算是用37Cl 信号的面积除以35Cl 信号的面积。结果表明,由于对背景信号的变化和噪声不敏感,线性回归斜率法具有更高的测试精度,对10 个CH3Cl 样品序列进行线性回归分析的仪器精密度为0.1‰,积分方法的精密度为0.15‰。为进一步评价该方法,研究还通过改变参数计算了具有较大拖尾的瞬态信号的同位素比值(改变纳入计算范围的色谱峰末端的位置,纳入计算范围的色谱峰前端的位置保持不变)。结果表明,即使在最极端的情况下仅使用了一半的信号,采用线性回归斜率法也未使同位素比值改变超过±0.15‰。
目前已有许多研究采用线性回归斜率法处理瞬态信号并得到了较好的结果。Epov 等[20]采用GC/MC-ICP-MS 测定汞形态同位素比值时,通过线性回归斜率法处理瞬态信号提高了准确性和精度(2SD=0.2‰~0.5‰)。Martinez等[21]通过LC/MC-ICP-MS在线分离测定水样中的亚硫酸盐、硫酸盐和硫代硫酸盐的δ34S时,用线性回归斜率法计算同位素比值,得到进样1 μg硫时,δ34S的不确定度小于0.25‰,重现性小于0.5‰。通过计算表明,选择斜率标准差最小化来定义线性回归斜率法计算的峰值区域提高了精度,并显著降低了不确定性。Kümmel 等[22]采用中分辨和低分辨率模式的GC/MC-ICPMS 实现了有机化合物特定形态中δ33S 和δ34S 的同时分析。采用线性回归斜率法计算同位素比值,结果表明进样量大于100 pmol 硫时,δ33S 和δ34S 的分析不确定度通常优于± 0.2‰(σ)。Sanabria-Ortega等[13]通过GC/MC-ICP-MS联用在线分离测定了铅同位素比值,同样发现采用线性回归斜率法得到了较好的计算精度和准确度。当进样1.2 ng 乙基化后的SRM NIST 981 铅溶液时,对于208/206Pb 和207/206Pb,精度(2RSDEXT,n=21)分别为49、69 ppm,实验结果与参考值的偏差均优于0.003 3‰和0.000 7‰。
此外,通过调整色谱峰的形状可以有效提高同位素分析的精度。Garciá-Ruiz等[23]使用IC与MCICP-MS 联用测定锶同位素比值时,通过调节进样量、样品和流动相的基体性质,得到了铷和锶2 种元素的信号均为平顶峰,发现在获得的平顶峰平台上有1 个稳定信号,可在3 min 内测量同位素比值。通过逐点计算法可以获得很好的精度。但是,并非所有元素的色谱分离都可以得到很好的平顶峰。
现有研究表明,采用全峰积分法时,需要保留色谱峰的整个峰尾直至背景上没有可识别的信号,才可最大程度地确保精度和准确度。另外,在采用全峰积分法时,减去背景基线时会引入误差,而这种误差在小信号(如峰尾端)时尤为明显。采用峰中心积分法可能比全峰积分法得到更好的同位素测试精密度,但得到准确同位素比值需要色谱分馏是围绕峰中心对称分布的。由于受到瞬态信号宽度的限制,逐点计算法受到可用于峰内平均同位素比值的点数的限制,尽管通过扩大色谱峰的宽度来增加点的数量可以提高测试精密度,但该方法会导致信号强度降低,甚至会导致不同元素或者形态色谱峰分辨率的降低从而影响形态同位素比值的测定。线性回归斜率法对背景信号的变化和噪声不敏感,对最密集的点赋予了更高的权重。采用线性回归斜率法时,较窄的峰的峰值灵敏度更高,积分时间更短,数据点数量增加,可获得更好的外部再现性。通过选择斜率标准差最小化来定义线性回归斜率法计算的峰值区域可以提高精度,并显著降低综合不确定性。因此,目前越来越多的研究更倾向于采用线性回归斜率法处理瞬态信号。
2 质量歧视校正
在使用MC-ICP-MS 测定元素同位素比值的过程中,仪器分析过程会引起不必要的同位素分馏,即质量歧视效应。例如碰撞散射[24]和空间电荷效应[25]可能会造成质量歧视效应,从而严重地影响同位素测定精度。传统的校正质量歧视效应的方法主要是内标法(Elemental doping)、标准-样品交叉法(Standard sample bracketing,SSB)和双稀释剂法(Double spike,DS)。
色谱与MC-ICP-MS联用在线分析同位素组成时,一方面除仪器自身的质量歧视效应外,色谱分离过程中同样可能会发生同位素分馏;另一方面,由于采集的是瞬态信号,法拉第前置放大器的响应速度会难以准确记录离子流的快速变化,因此同样可能会造成同位素比值的漂移[26]。色谱与MCICP-MS联用研究中,研究人员陆续发展了不同的质量歧视校正方法来校正同位素比值漂移。
Guéguen等[27]用LC/MC-ICP-MS测量核应用中的钕同位素比值时,发展了内注射标准-样品交叉法(IISSB),以在Nd 洗脱峰前后引入参考标准品为基础进行质量偏差校正。由于在辐照样品中钕的比例不固定,所以无法使用自身的同位素比值校正质量歧视效应,因此研究者采用SSB 法以消除基质效应,提出必须在与样品相同的条件下通过LC系统进样;而且为了获得最佳性能,要求样本和两个标准之间的分析时间很短。在开发的IISSB 方法中,为优化交叉标准注射和梯度匹配,研究者设计了一种“双进样LC/MC-ICP-MS”特殊装置(如图3),保证了样品和标准之间的基质匹配。所得Nd测试结果与采用离线分离、SSB法得到的结果相当。
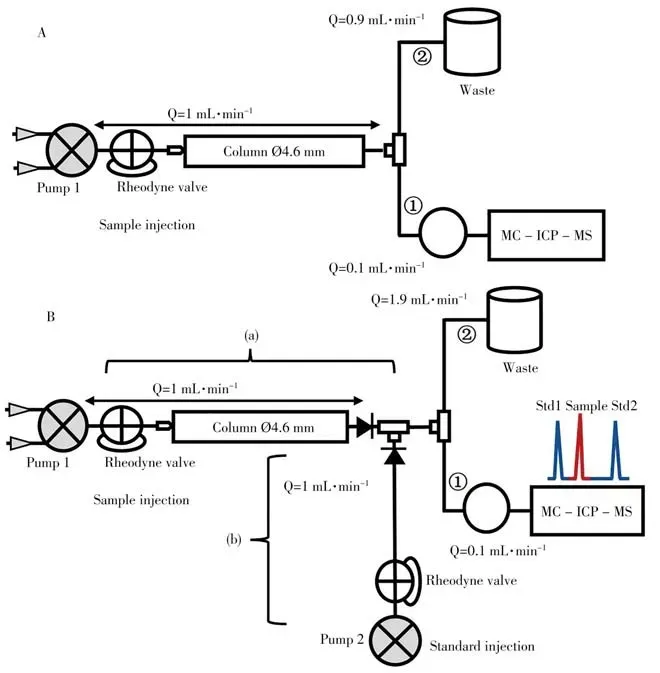
图3 LC/MC-ICP-MS系统(A)与双进样LC/MC-ICP-MS系统(B)[27]Fig.3 LC/MC-ICP-MS system(A)and the dual inlet LC/MC-ICP-MS system(B)[27]
用MC-ICP-MS在瞬态信号中测定同位素比值时,通常在信号采集过程中会发生同位素信号比值的漂移。这种“同位素漂移”与法拉第探测器配置中相关的放大器响应时间滞后(尽管比较小,但各法拉第杯响应时间会有明显差异)有关。Gourgiotis 等[28]提出了一种将瞬态同位素信号同步的方法—内部信号同步法(MISS),使用原始同位素信号的比值量化放大器之间的时间滞后,校正了同位素比值漂移。采用LC/MC-ICP-MS 分析Nd 同位素比值时,同位素漂移主要来自LC 和检测系统两个部分,Gourgiotis 等[26]首先利用MC-ICP-MS 放大器的时间常数成功校正了检测系统产生的同位素漂移,校正后,可以明显看到来自LC 的漂移。进一步地,使用MISS 有效地校正色谱漂移和计算钕同位素之间的时间滞后(0.003 6 s/amu)。根据漂移校正数据(色谱和电子漂移校正)计算出6 次注射的Nd 比值,并与Guéguen 等在未进行漂移校正情况下获得的结果(原始数据)进行比较。经MISS 校正后的142Nd/144Nd、146Nd/144Nd 和150Nd/144Nd 的重复性分别提高了1.5、1.7 和2.7 倍,但对于其余的比值,MISS 未产生改善效果。值得说明的是,MISS可以显著提高单次进样的同位素比值的不确定性,而不是比值的重复性。
Martinez等[21]通过LC/MC-ICP-MS在线分离测定水样中亚硫酸盐、硫酸盐和硫代硫酸盐的δ34S时,提出了一种基于样品中直接添加内标三甲基氯化亚砜(TMSO)的新方法(IS)来校正质量歧视效应。图4显示了由元素分析仪-同位素比值质谱仪(EA-IRMS)测量的所有分析溶液中不同物质的δ34S值,以及用LC/MC-ICP-MS采用不同校正方法(IS、CUB、ISEC、CSB)得到的δ34S值。其中,CUB是指分别在每个样本之前和之后进行的TMSO测量的标准-样品交叉法;ISEC是指首先用IS方法计算,并在整个分析过程中分析每个阴离子的标准,建立每个阴离子的校准曲线,然后用于校正δ34S值;CSB是指标准与样品是相同的阴离子的标准-样品交叉法。结果表明,当使用ISEC时,质量偏差和柱上同位素分馏分别通过在样品中加入内标和外标法进行了校正,ISEC是同时测定亚硫酸盐、硫酸盐和硫代硫酸盐中δ34S的最准确和稳健的方法。
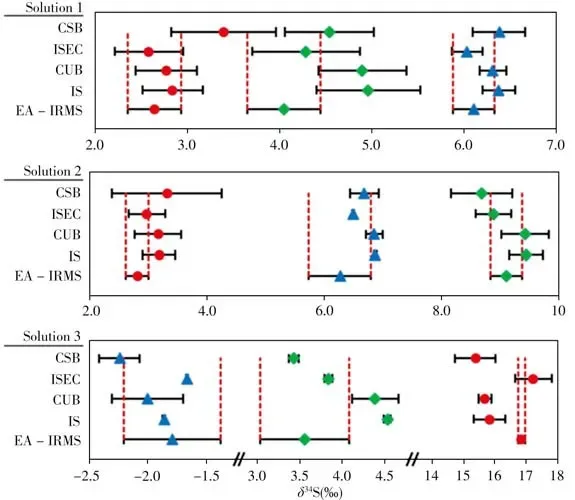
图4 对比EA-IRMS(50 μg S,n ≥2)和LC/MC-ICP-MS(1 μg S)测定混合标准溶液1、2和3中不同物质的δ34S[21]Fig.4 δ34S of different substances in standard solutions 1,2 and 3 measured by EA-IRMS(50 μg S,n ≥2)and LC/MC-ICP-MS(1 μg S)[21]
色谱与MC-ICP-MS联用在线分离测定同位素比值时,因样品和标样的浓度和基体不匹配、色谱分离时发生同位素分馏、采集瞬态信号产生的同位素漂移等问题,采用传统的内标法和标准-样品交叉法来校正质量歧视效应通常无法直接满足色谱联用时的同位素测试要求。从现有研究来看,根据不同的测试条件,往往需要在现有基础上改进和发展新的校正方法(如IISSB、ISEC、MISS 等)以获得准确的同位素比值。
3 典型应用
3.1 卤素
含氯脂肪族碳氢化合物(CAHs),如三氯乙烯(TCE)和四氯乙烯(PCE),是广泛存在的地下水污染物,且会保留相对较长的时间。通过碳和氯的稳定同位素组成可以表征CAHs 的降解情况。Van Acker等[15]采用GC/MC-ICP-MS 成功分离并测定了模拟CAHs 样品中TCE 和PCE 的氯同位素比值,含24~165 μg 氯的样品的δ37Cl 测量精度为0.12‰(2SE)。虽然该方法的精确度不如稳定同位素比值质谱,但具有快速分析多组分混合物中每种CAHs的能力,且降低了样品制备的复杂性,大大缩减了分析时间,具有对环境污染样品进行Cl同位素分析的潜力。Horst等[19]使用GC/MC-ICP-MS同样分离测定了实验室模拟样品的挥发性脂肪族化合物中的不同形态氯同位素比值,得到了与传统的离线双入口同位素比值质谱仪(DI-IRMS)一致的结果。这种简单的方法适用于挥发性脂肪族化合物的环境和实验室样品,例如来自受污染含水层的环境样品,从而表征有机化合物的来源、转化途径等。Renpenning 等[29]建立了GC/MC-ICP-MS测定半挥发性有机物中不同形态37Cl/35Cl值的方法,进一步扩大了可分析化合物的范围,混合物的分析精度通常优于±0.3‰,准确度在±0.2‰以内。Lihl 等[30]在合成氯形态同位素标准物质时,利用GC/MC-ICP-MS测定氯同位素比值。
Wu等[31]利用开发的GC/MC-ICP-MS技术研究了食物网中六氯化苯(HCH)的转化反应。从3个不同的污染地点采集了HCH污染的土壤、植物、牛/水牛奶和粪便、野生动物肝脏和海豹脂肪进行分析,测定α-HCH和β-HCH的同位素组成,以分析与潜在食物网反应运输相关的转化过程。发现α-HCH的同位素组成δ37Cl 变化范围为-0.86‰~4.33‰,变化高达5.19‰;β-HCH 的同位素组成δ37Cl 变化范围为-2.19‰~4.21‰,变化高达6.40‰。作为来源的3个HCH淤泥样品中δ37Cl的同位素组成的平均值分别为-1.38‰±0.57‰(α-HCH)和-1.27‰ ±0.69‰(β-HCH)。与来源相比,土壤和植物对α-HCH 的δ37Cl最大富集达2.54‰,表明植物对HCH的吸收会导致Cl的同位素分馏,这可能是根际生物降解或植物体内生物降解的结果。与文献报道的HCH来源的同位素组成相比,在牛乳、牛粪和海豹脂肪中获得的HCH同位素值比文献报道的来源范围上限更大(δ37Cl为3.79‰)。粪便和牛奶中有强烈的同位素富集,表明HCH在牛/水牛代谢过程中可能在消化道发生降解。牛奶、肝脏和海豹脂肪中HCH的显著同位素富集进一步表明,在高强度的代谢后,只有一部分残留在脂肪中积累。HCH残留组分的同位素富集反映了高等生物的代谢降解改变了生物体内HCH的浓度,因此残留部分的浓度不能充分代表生物暴露。
溴同位素组成对环境中有机溴的降解监测和源解析具有潜在的诊断价值。Holmstrand 等[32]将GC/MC-ICP-MS 用于含2,2′,4,4′-四溴联苯醚(BDE-47)和甲氧基-2,2′,4,4′-四溴联苯醚(MeO-BDE-47)的环境样品中溴同位素比值的测定,该样本是从一只搁浅在瑞典西海岸的突吻鲸的鲸脂中提取。使用BDE-47 组分作为同位素参考,在相隔1 个月的2 次分析中,同位素组成(Δ81Br)的平均差异为-0.3%±0.7%(1 s,n=6)。BDE-47 和MeO-BDE-47 在δ81Br 上的差异可以忽略,表明BDE-47和MeO-BDE-47 的形成机制非常相似。因此,这两种化合物可能有相似的来源,或是同位素效应的差异(如果有不同的形成途径)小于目前可以解析的。
3.2 金属元素
汞是一种全球性污染物,其生物地球化学循环与形态密切相关,如汞能以Hg0的形态从水生系统挥发到大气中;以Hg(Ⅱ)的形态从大气中沉降;在水环境中甲基化生成有毒的甲基汞;由于甲基汞在生物链中有更大的营养转移效率,汞在食物链中可以产生强的生物积累效应等[33-34]。Epov 等[33]采用GC/MC-ICP-MS同时测定了不同汞形态的同位素组成,获得的δ202Hg外精度2SD为0.56‰,这是首次报道的能够测定实际样品中特定形态汞稳定同位素组成的分析方法。研究者采用GC/MC-ICP-MS 测量了3种不同样品的汞同位素比值:标准物质BCR-CRM-464(金枪鱼)、标准物质IAEA-085(人发)和二次参比标准UM-Almadén。根据测量结果,发现BCR-CRM-464(金枪鱼)偶数同位素符合质量分馏(MDF)规律,而奇数同位素有强烈的非质量分馏(MIF)。Rodríguez-González 等[34]采用GC/MCICP-MS 法,首次报道了厌氧细菌在黑暗条件下Hg(Ⅱ)甲基化过程中汞的稳定同位素分馏。结果表明,在黑暗条件、厌氧细菌存在情况下,Hg(Ⅱ)的甲基化导致Hg(Ⅱ)底物和产生的单甲基汞(MMHg)的汞同位素的MDF。这种过程在细菌的指数增长下发生,发现这种细菌优先甲基化汞的轻同位素。在连续培养96 h,单次采样培养140 h后,在相同的细胞密度下观察到样品分馏趋势的变化,说明同时平衡甲基化程度(如去甲基化)的过程的贡献增加。
铅同位素在地质年代学、污染来源示踪等研究中应用日益广泛,也有学者开展了与色谱联用快速分析铅同位素的研究。Sanabria-Ortega 等[13]提出了一种GC/MC-ICP-MS 在线分离测定复杂基质中铅同位素比值的方法,并应用于沥青质、原油和干酪根样品中的铅同位素分析。样品经酸消解后,铅被乙基化为PbEt4,以异辛烷萃取分离后用GC/MC-ICP-MS 进行分析。测定结果与传统过柱纯化后在“干”和“湿”等离子体条件下得到的结果有很好的一致性。与传统方法相比,该方法的主要优点为:①样品制备简单,制备和测量时间缩短了15倍;②无需样品蒸发而实现了样品预浓缩,减少了交叉污染;③降低高纯试剂(如酸)的消耗量,从而降低操作成本。Penanes等[35]采用LC/MC-ICP-MS实现了考古材料中铅同位素比值的直接测定,不需要传统的离线分离富集纯化步骤。但该方法仅适用于铅含量高于500 μg/g的样品,对于低浓度样品仍需要预浓缩。
锶同位素常被用作示踪剂应用于环境样品、食物样品等的溯源研究。Garciá-Ruiz 等[23]采用IC 与MC-ICP-MS联用,在线分离干扰元素并测定锶同位素比值。该方法适用于广泛样品基质中锶同位素的测定,已被应用于苹果酒、土壤渗滤液、苹果叶和苹果样品,以研究锶同位素比值在土壤-树-苹果酒系统中的迁移[36]。Rodríguez-Castrillón 等[37]采用离子交换色谱与MC-ICP-MS联用在线分离测定锶同位素比值,在锶含量为50 ng/g 时得到88/87Sr 的精度为0.006%~0.010%,并使用该方法成功测定了英格兰和西班牙的苹果酒样品中的锶同位素比值。
由于锑的广泛使用和锑的开采及冶炼活动,大量锑被释放到环境中,造成了严重的锑污染。因此,锑同位素在环境系统中的研究也日益受到重视[38]。Wehmeier 等[17]采用GC/MC-ICP-MS 测定三甲基锑中的锑同位素比值,测定了来自实验室污水污泥发酵罐的真实样本,并观察到生物产生的三甲基锑的同位素分馏(δ123Sb+10‰和+19‰),说明当锑被厌氧细菌甲基化时会发生同位素分馏。
上述结果表明,色谱与MC-ICP-MS联用进行金属同位素的研究还较少,而且主要集中在同位素比值差异较大的放射性成因的同位素分析,这主要是因为金属稳定同位素在自然样品中的分馏较小,而联用形态同位素分析技术提供的同位素测试精度仍需进一步改善以满足实际需求。
3.3 硫
硫是一种重要的微量元素,广泛参与生物地球化学过程,而且在某些污染物的生物降解中发挥重要作用。因此,测量不同化合物或环境样品中硫同位素组成对于了解硫循环,研究与硫氧化还原反应相关的同位素分馏非常有用。Santamaria-Fernandez等[39]发展了通过HPLC/MC-ICP-MS测量S同位素来鉴别1种抗病毒药物真伪的方法。研究人员分析了从制药合作方收集的417片药(每批3片,n=139),测试得到每批药片的平均δ34S。结果发现大多数药片(n= 96)的δ34S = 3.6‰,不确定度为1‰(n= 96,k=2),这些药片被确认为正品。但部分药片(n=23)的δ34S与之显著不同,可能是假药。
此外,色谱与MC-ICP-MS 联用技术还可用于同位素示踪的研究中。San Blas 等[40-41]采用HPLC/MC-ICP-MS在线分离测定了小鼠口服34S标记酵母后尿液中的不同硫代谢物的硫同位素比值,通过对健康小鼠和前列腺癌小鼠的尿液代谢物进行硫同位素分析开展了硫代谢研究。研究者分别分析了尿液中总的硫同位素组成和不同代谢物中的硫同位素组成,发现使用富含34S的酵母,测量示踪或被示踪物同位素比值可以用于区分健康小鼠和前列腺癌小鼠。结果表明,硫同位素富集程度不随疾病的进展而增加,因此在前列腺癌的第一阶段,患有前列腺癌的小鼠可与健康的小鼠区分开。Ullrich 等[42]采用IC/MC-ICP-MS 进行硫同位素分析,观测硫代砷酸盐([HAsVSn-IIO4-n]2-,n=1~4)在非生物氧化过程中的分馏。应用此方法发现,一硫代砷酸盐在氧化过程中与产物硫酸盐相比,分馏高达6.1‰;由于与硫化物分子间同位素交换,一硫代砷酸盐的硫同位素富集可达9.1‰。而四硫代砷酸盐通过三硫代砷酸盐和二硫代砷酸盐氧化成一硫代砷酸盐并未导致分馏。这些结果有助于阐明硫代砷酸盐转化的途径,从而为硫环境同位素分馏模式的解释提供了有价值的信息。Martinez等[21]采用LC/MC-ICP-MS在线分离测定了水样中的亚硫酸盐、硫酸盐和硫代硫酸盐的δ34S,当注入1 μg 硫时综合不确定度小于0.25‰,再现性低于0.5‰。该方法可应用于大多数的环境水样,可作为研究形成亚硫酸盐和硫代硫酸盐等中间价硫阴离子的硫氧化还原过程的有力工具。Faβbender等[43]建立了CE与MC-ICP-MS 联用在线硫同位素分析方法,测定硫酸盐δ34S 值的精度为2SD=0.3‰~1.3‰。而IRMS 对位于亚洲、欧洲和北美的许多不同河流系统的河水中硫酸盐的硫同位素分析结果表明,δ34S值在-4‰~+18‰之间,因而使用CE/MC-ICP-MS 足以揭示不同河流系统中硫酸盐-δ34S 的差异。Kümmel 等[22]采用中分辨和低质量分辨率模式的GC/MC-ICP-MS实现了有机化合物特定形态中δ33S和δ34S的同时分析,并将该方法应用于工业生产的有机化合物的δ33S和δ34S同位素分析。对4种工业生产的有机化合物(噻吩(THI)、四氢噻吩(THT)、二乙基硫醚(DES)和二甲基二硫醚(DMDS))的混合物进行了测量,这些化合物的δ33S 和δ34S 之间具有质量分馏趋势(Δ33S ≤±0.2‰)。将DMDS 暴露于紫外辐射3 h 引起光解降解并测定了非质量分馏(MIF),结果表明通过直接光解降解二甲基二硫醚会产生微小但显著的MIF(Δ33S = 0.55‰ ±0.04‰,n=3),表明该方法对这些类型的研究具有足够的灵敏度。
3.4 其他
色谱与MC-ICP-MS 联用方法同样被应用于核材料的分析。Günther-Leopold 等[44]用HPLC/MCICP-MS 分离干扰元素,在线测定了核燃料样品中的钚同位素比值。外部重现性为0.04%~0.2%(2SD),且无需繁琐的样品前处理过程(尤其是对于需特别预防的放射性物质)。Caruso等[45]采用HPLC/MC-ICP-MS 分别测定了4 根辐照燃料棒中的Gs、Eu 同位素,大多数样品的同位素测试统计误差在0.4%~0.8%(1σ)范围内。Guéguen 等[27,46]采用LC/MC-ICP-MS 测定了核样品中的Gd、Eu、Sm、Nd同位素比值,并对模拟样品进行分析验证了该方法的可行性,几乎所有同位素比值的重现性均优于2‰(k=2)。Martelat等[47-48]采用CE/MC-ICP-MS分别实现了在线Nd、U、Pu同位素比值的测定。
硅同位素作为生物地球化学示踪剂,已被用于研究自然界的各种过程,如风化和生物过程,提高了人们对全球硅生物地球化学循环的认识。Yang 等[49]采用离子排阻色谱(IEC)与MC-ICP-MS 耦合在线测定了天然水体和几种商业硅标准中的硅同位素比值,发现硅同位素比值不仅在几种商业硅标准中存在显著差异,而且在天然水体中也存在显著差异。Zakon等[50]采用GC/MC-ICP-MS测定了有机化合物中的碳同位素比值δ13C,对于含有低至0.6 nmol 碳的样品其精度为0.3‰(1σ),验证了GC/MCICP-MS 联用技术可用于有机化合物单体中δ13C 的精确分析。Karasinski 等[16]采用IC/MC-ICP-MS 分别在湿和干等离子体条件下精确测定了镁同位素比值,对富基质天然水和岩石样品进行了镁同位素分析。湿等离子体和干等离子体条件下的镁同位素比值精度均很好,一般为0.15‰(2SD),两者均可与离线Mg分离和连续测量相媲美。
4 结 论
色谱与MC-ICP-MS 联用测定同位素比值,不仅具有MC-ICP-MS 测定同位素比值的优势,而且由于色谱的分离作用,可以:①将待测元素与干扰离子和基体有效在线分离,大大缩减了样品预处理时间,有望提高同位素测试的效率和改善分析自动化水平;②将同一元素的不同形态分离开,测定特定元素形态的同位素比值,从而获取比总量同位素比值更丰富、更有效的信息;③使同时分析多个元素形态的同位素比值成为可能。
色谱与MC-ICP-MS联用在线分析的主要挑战是瞬态信号的处理和分析过程中的质量歧视校正。大量文献表明,线性回归斜率法是处理瞬态信号计算同位素比值的最好方法。根据不同的测试条件,通常需在内标法和标准-样品交叉法的基础上发展新的校正方法(如IISSB、ISEC、MISS等)来校正质量歧视效应。
目前,采用色谱与MC-ICP-MS联用在线分离测定同位素比值的研究越来越多。但形态同位素分析的技术还不是十分成熟,分析精度仍需进一步提高。例如Entwisle 等[51]采用高效液相色谱和冷蒸气发生多接收电感耦合等离子体质谱(CVG/MC-ICP-MS)联用测定了鱼组织中甲基汞的汞同位素比值,对在线耦合和离线测量进行了比较和评估,发现离线测量是更加稳定且可重复的分析方法。因此,仍需要进一步完善分析方法,找到更好的瞬态信号处理和校正质量歧视效应的方法,克服流动相基体对同位素测试的影响以及流速匹配的问题,进一步提高分析速度和同位素测试精度,使其能发挥更大的作用。一方面,需要建立更有效的显著降低色谱同位素分馏的形态分离方法;另一方面,色谱在线同位素分析对接口以及MC-ICP-MS质谱仪器的性能同样有了更高的要求,迫切需要提高样品的利用效率以及检测灵敏度来得到更理想的同位素分析精度。总之,通过形态同位素分析,可以了解不同元素形态的生物地球化学循环过程,为解决各种生命和环境问题提供更有效的信息,因此形态同位素分析在地球科学、环境、生命科学以及化学等领域将会发挥越来越重要的作用,但其广泛应用还有赖于可形态分析的同位素体系的扩展与分析方法性能的有效提升。
