不同晶型MnO2纳米阵列的可控合成及其电催化析氧性能
2022-02-17黄乐珩赵英霞谢启星唐诗昌
黄乐珩 程 高,2,3 赵英霞 谢启星 孙 钺 唐诗昌 孙 明,2,3 余 林*,,2,3
(1广东工业大学轻工化工学院,广东省教育厅清洁化学技术重点实验室,广州 510006)
(2广东工业大学轻工化工学院,广东省植物资源生物炼制重点实验室,广州 510006)
(3广东工业大学轻工化工学院,广州市清洁交通能源化学重点实验室,广州 510006)
0 引 言
利用电解水技术高效制备氢气被认为是解决全球能源危机的主要途径之一[1]。析氧反应(OER)作为电解水的关键半反应,涉及O—H键的断裂、O—O键的形成和4H+/4e−的多步转移等复杂反应,需要较高的能量来克服动力学壁垒,这严重阻碍电解水的能量转换效率[2]。目前已商用的贵金属基材料(如IrO2、RuO2等)对OER展现出卓越的电催化性能,但是成本高和耐久性差是其广泛应用的主要障碍[3]。因此,探索低成本、高效稳定的非贵金属基催化剂材料,对提高电解水能量转化效率具有重要的现实意义。过渡金属基催化剂(如过渡金属氧化物[4]、氢氧化物[5]以及它们的复合材料[6-8]等)成本低、OER活性和稳定性良好,被认为是具有前景的可替代贵金属催化剂的催化材料之一。
二氧化锰(MnO2)价格低廉,晶体结构丰富,具有较好的电催化活性和稳定性,在OER电催化方面展现出巨大的应用潜力[9]。MnO2具有诸多晶型,如α、β、δ、γ等[10-13],其晶体结构与OER电催化活性存在一定的相关性。例如,Suib等[14]发现OER的催化性能高度依赖于MnO2晶体结构,OER的催化活性遵循以下顺序:α-MnO2>amorphous MnO2>β-MnO2>δ-MnO2。Pala等[15]合成不同晶型的MnO2纳米结构,发现其每电化学表面积单位的OER活性依次为δ-MnO2>α-MnO2>γ-MnO2>β-MnO2>r-MnO2。上述报道中MnO2晶型与OER活性之间的规律不完全相同,这可能是MnO2制备方法和原料的差异所致。因此,采用相同的制备方法,通过改变反应参数制备不同晶型的MnO2,并系统研究MnO2的晶型结构与其OER催化活性之间的规律仍然具有重要的研究意义。
用于OER的粉末型催化剂电极通常是通过添加聚合物黏合剂和导电炭黑,在导电载体上涂覆粉末催化剂来制备。这种情况下,粉末催化剂的活性位点未得到充分暴露,并且在大电流和含氧量高的环境下,黏结剂降解,电极表面的粉末催化剂容易随着气泡的释放而脱落,从而降低OER催化活性[16]。与粉末型催化剂相比,自支撑阵列电催化剂直接生长在导电载体上,具有更牢固的纳米阵列结构。阵列结构使活性位点充分暴露,提高导电性,加快电子传输,促进OER过程中OH−的吸附和O2的析出[17-18],呈现出高活性和耐久性。例如,Co2FeO4纳米片阵列[19]、Co掺杂CuO纳米阵列[20]、Co3O4@NiMn-LDH三维异质结构阵列[17]等等,均可表现出优异的OER催化性能。目前,有不少研究报道了MnO2纳米阵列,包括 α-MnO2纳米线[21]、β-MnO2纳米棒[22]、δ-MnO2纳米片[23]、γ-MnO2纳米棒[24]等。迄今为止,在导电载体上可控合成不同晶型的MnO2纳米阵列的报道仍然较少[25-26]。此外,这些报道所采用的合成条件较苛刻,如水热时间长、高温煅烧等。因此,通过采用温和的制备方法,在导电载体上可控合成不同晶型的MnO2纳米阵列,并进一步将其应用于电催化析氧仍有待研究。
基于上述报道,我们采用水热法,改变反应温度和硫酸的用量,在碳纸(CFP)基底上分别合成了α-MnO2纳米线和δ-MnO2纳米片阵列,并考察在碱性介质中的OER催化性能。CFP拥有优良的导电性能和稳定性,适合作为OER支撑基底负载活性材料,不仅有助于改善MnO2导电性和团聚现象,而且有利于简化电极制备过程。在碱性介质中,α-MnO2(10 mA·cm−2处的过电位为444 mV)的OER电催化活性明显优于 δ-MnO2(10 mA·cm−2处的过电位为 522 mV),这是不同晶型MnO2的表面氧空位浓度和Mn3+含量不同导致的。
1 实验部分
1.1 材料与试剂
CFP、盐酸、无水乙醇、高锰酸钾、无水亚硫酸钠、98%浓硫酸均为分析纯,未经处理,直接用于反应。实验用水是去离子水。
1.2 实验方法
CFP预处理:将商用CFP(约1 cm×4 cm)依次用丙酮、盐酸溶液(3 mol·L−1)、无水乙醇和去离子水超声清洗处理15 min后,放置在60℃的烘箱内干燥12 h。
α-MnO2纳米线阵列:分别称取0.158 1 g的高锰酸钾和0.189 1 g的无水亚硫酸钠,溶于30 mL去离子水中,并加入20µL的浓硫酸,在室温下磁力搅拌30 min后形成澄清的紫红色溶液。将混合溶液转移到50 mL的聚四氟乙烯内衬的反应釜中,随后将处理完的CFP插入反应釜内。将密封好的不锈钢反应釜置于180℃的烘箱中恒温反应8 h。待反应结束并自然冷却至室温后,可以获得表面沉积一层褐色沉淀物的CFP,然后分别用去离子水和无水乙醇将其反复超声清洗后,在60℃下干燥12 h,得到生长在CFP上的α-MnO2。α-MnO2在CFP上的负载量约为1.63 mg·cm−2。
δ-MnO2纳米片阵列:分别称取0.158 1 g的高锰酸钾和0.189 1 g的无水亚硫酸钠,溶于30 mL去离子水中,在室温下磁力搅拌30 min后形成澄清的紫红色溶液。将混合溶液转移到50 mL的聚四氟乙烯内衬的反应釜中,随后将处理完的CFP插入反应釜内。将密封好的不锈钢反应釜置于150℃的烘箱中恒温反应8 h。待反应结束并自然冷却至室温后,可以获得表面沉积一层褐色沉淀物的CFP,然后分别用去离子水和无水乙醇将其反复超声清洗,之后在60℃下干燥12 h,得到生长在CFP上的δ-MnO2。δ-MnO2在CFP上的负载量约为1.54 mg·cm−2。
采用以上的合成步骤进行了对照实验:分别在其他条件不变的情况下150℃添加20µL的浓硫酸和180℃不添加浓硫酸进行水热反应,目的是考察反应温度和硫酸加入量对MnO2产物的影响。
1.3 材料结构表征
样品的物相分析采用X射线衍射仪(XRD,Bruker D8,荷兰PAN alytical),Cu Kα射线,λ=0.154 16 nm,管电压为40 kV,管电流为40 mA,扫描范围2θ=10°~80°;样品的微观形貌观察采用场发射扫描电子显微镜(SEM,SU 8020,Hitachi,加速电压5 kV)、透射电子显微镜(TEM,Talos F200s,场发射电压200 kV);样品表面的元素及元素化合价态采用X射线光电子能谱仪(XPS,Escalab 250Xi,英国 Thermo Fisher),X射线源为单色化Al Kα(1 486.6 eV),束斑为400µm。
1.4 电化学性能测试
采用德国Zahner电化学工作站的三电极体系进行电化学性能测试,电解液为1.0 mol·L−1KOH溶液,工作电极为所制得电极(测试面积约为1 cm2),参比电极为3.5 mol·L−1KCl的Ag/AgCl电极,对电极为碳棒。极化(LSV)曲线的扫描速率为5 mV·s−1,电压范围为0.1~0.8 V(vs Ag/AgCl)。电化学阻抗谱(EIS)测试在0.55 V(vs Ag/AgCl)下进行,频率范围为0.1~10 000 Hz。稳定性测试的电流密度为10 mA·cm−2,时间为12 h。以上电化学测试的电位需校正为相对可逆氢电极(RHE)的电位,计算公式:
E(vs RHE)=E(vs Ag/AgCl)+0.059pH+0.204 6
2 结果与讨论
2.1 催化剂的结构与形貌分析
分别对在150℃、0µL浓硫酸和180℃、20µL浓硫酸条件下制备的产物进行XRD表征(图1a)。图1b、1c分别为α-MnO2和δ-MnO2的结构示意图。以KMnO4作为氧化剂,Na2SO3作为还原剂,当温度为180℃且添加浓硫酸时,所得样品的衍射峰信号与α-MnO2(PDF No.44-0141)标准卡片一一对应,表明此产物是α-MnO2。在150℃、不添加浓硫酸的条件下所制备样品的XRD图在12.3°、24.8°、37.4°和65.4°处有明显的衍射峰,分别对应δ-MnO2(PDF No.86-0666)的(003)、(006)、(012)和(110)晶面,说明在该条件下的产物是δ-MnO2。δ-MnO2的结晶度较差,可能是由于较低的结晶温度和成核速率阻碍了[MnO6]八面体的长程有序性[27]。以上XRD结果表明在CFP上分别成功地生长了α-MnO2和δ-MnO2纳米阵列,MnO2的晶型与反应条件密切相关。
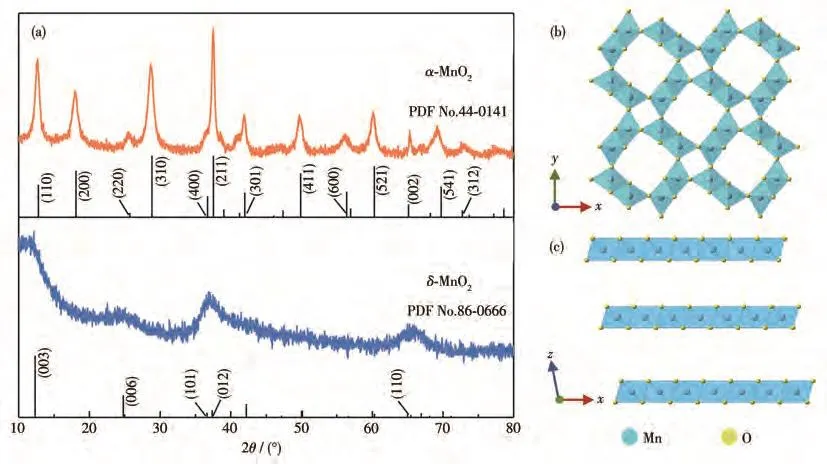
图1 (a)α-MnO2和δ-MnO2的XRD图;(b)α-MnO2和(c)δ-MnO2的结构示意图Fig.1 (a)XRD patterns of α-MnO2 and δ-MnO2;Structural illustrations of(b)α-MnO2 and(c)δ-MnO2
图2a、2b和 2d、2e分别为 α-MnO2和δ-MnO2的SEM图。通过低倍图可清楚观察到,α-MnO2纳米阵列由沿随机方向生长的纳米线组成,δ-MnO2纳米阵列由不规则的二维纳米片组成。纳米线和纳米片几乎垂直排列,紧密而均匀地负载在碳纤维管上,其中一些纳米片之间相互交连,一些纳米线的末端相互交叉,连接形成了一个网状结构,这种结构能有效增大催化材料与电解液的接触面积,缩短电解质离子的扩散距离,提高OER过程中的物质传输效率[28-30]。通过TEM和HRTEM(高倍TEM)进一步探测MnO2的微观形貌。图2c和2f分别为α-MnO2和δ-MnO2的TEM和HRTEM图。在HRTEM中可看出α-MnO2形貌为纳米线,直径为12~14 nm,长度为0.8~1.3 µm,晶格间距为0.24 nm,对应α-MnO2的(400)晶面。图2f显示δ-MnO2纳米片边缘弯曲,晶面间距为0.57 nm,这是由于在电子束照射下,水钠锰矿型δ-MnO2会发生脱水,导致(003)晶面的晶格距离减小,低于原本层间距离(0.72 nm)[31-32]。上述的HRTEM的结果均能与XRD图的衍射峰对应,说明成功在CFP上合成α-MnO2和δ-MnO2。

图2 (a、b)α-MnO2和(d、e)δ-MnO2不同放大倍数的SEM图;(c)α-MnO2和(f)δ-MnO2的TEM和HRTEM图(插图)Fig.2 SEM images with different magnifications of(a,b)α-MnO2 and(d,e)δ-MnO2;TEM and HRTEM images(Inset)of(c)α-MnO2 and(f)δ-MnO2
2.2 合成参数对MnO2晶型和形貌的影响
水热反应条件往往会对产物的结构和形貌造成较大影响,因此在150℃、添加20µL的浓硫酸和180℃、不添加浓硫酸的条件下进行水热反应以作为对照组,研究反应温度和添加浓硫酸对产物结构和形貌的影响。
150℃水热条件下,当反应体系中不存在浓硫酸时,产物为 δ-MnO2纳米片(图 1c、2d)。当添加 20µL浓硫酸时,所得产物的XRD如图3a所示,虽然产物的主相仍为δ-MnO2相,但12.3°处衍射峰消失。从图3b的SEM图可见,部分纳米片发生溶解,出现少量α-MnO2纳米线,这表明增加反应体系中H+含量有利于α-MnO2的形成。反应温度升至180℃时(图3c),不添加浓硫酸条件下所得产物在12.8°、36.7°、42.0°和49.8°处演化出对应α-MnO2(PDF No.44-0141)的(110)、(400)、(301)和(411)晶面特征峰,25.0°、37.3°和 65.8°处仍为 δ-MnO2(PDF No.86-0666)的特征峰,说明主体结构仍是δ-MnO2相,此时SEM图显示产物纳米片结构与纳米线结构共存(图3d),这与此前报道中提高温度有利于δ-MnO2转变为α-MnO2的结果一致[33-34]。
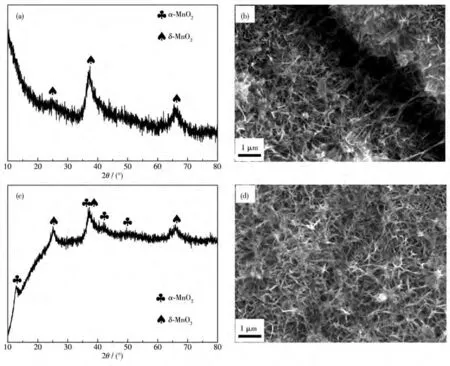
图3 不同条件下制备的MnO2产物的XRD图和SEM图Fig.3 XRD patterns and SEM images of MnO2 prepared at different reaction conditions
反应温度较低、不添加浓硫酸时,水热条件下[MnO6]八面体趋向于形成亚稳结构的δ-MnO2纳米片[35]。随着反应温度的提高和浓硫酸的加入,δ-MnO2发生溶解−重结晶过程,转化为热力学结构更稳定的α-MnO2,表现为δ-MnO2纳米片发生溶解,α-MnO2纳米线开始出现,直至完全转变为α-MnO2纳米线[34,36]。较高的反应温度有利于晶体的各向异性生长,因此高温下δ-MnO2纳米片亚稳结构发生卷曲,溶解再结晶形成长径比更高的α-MnO2纳米线结构[33]。高酸度对促进KMnO4和Na2SO3氧化还原反应动力学起着重要作用[36],添加浓硫酸促进反应平衡往正方向进行,同时随着浓硫酸的加入,有利于增加亚稳态二维结构δ-MnO2的溶解度,发生结构重组过程,进而使晶型转变为更稳定的一维结构α-MnO2[37]。
2.3 催化剂的电催化析氧性能
在氧气饱和的1 mol·L−1KOH溶液中进行电化学测试以评估OER活性。图4a为α-MnO2、δ-MnO2以5 mV·s−1扫描速率进行测试获得的LSV曲线。相较于 δ-MnO2(10 mA·cm−2处的过电位为 522 mV),α-MnO2具有更优的OER活性,其在10 mA·cm−2处的过电位为444 mV。通过LSV曲线计算得到相应的Tafel斜率(图4b),用于进一步研究OER的动力学。OER被认为是一个四步反应,每一步都伴随着电子转移。Tafel斜率的减小表明随着OER速率的提高,表面反应的决速步骤得到了改善。α-MnO2的Tafel斜率为 115.7 mV·dec−1,远小于 δ-MnO2(226.9 mV·dec−1),表明α-MnO2在催化OER过程中为涉及第一电子转移步骤和第二电子转移步骤的混合决速步骤,δ-MnO2的决速步骤为第一电子转移步骤,说明α-MnO2在OER过程中的反应速率更快,更有利于OER反应[38-40]。将制备的α-MnO2和δ-MnO2与近期文献报道的MnO2类电催化剂的OER活性进行对比(表1)可知,α-MnO2具有较为优异的OER活性。
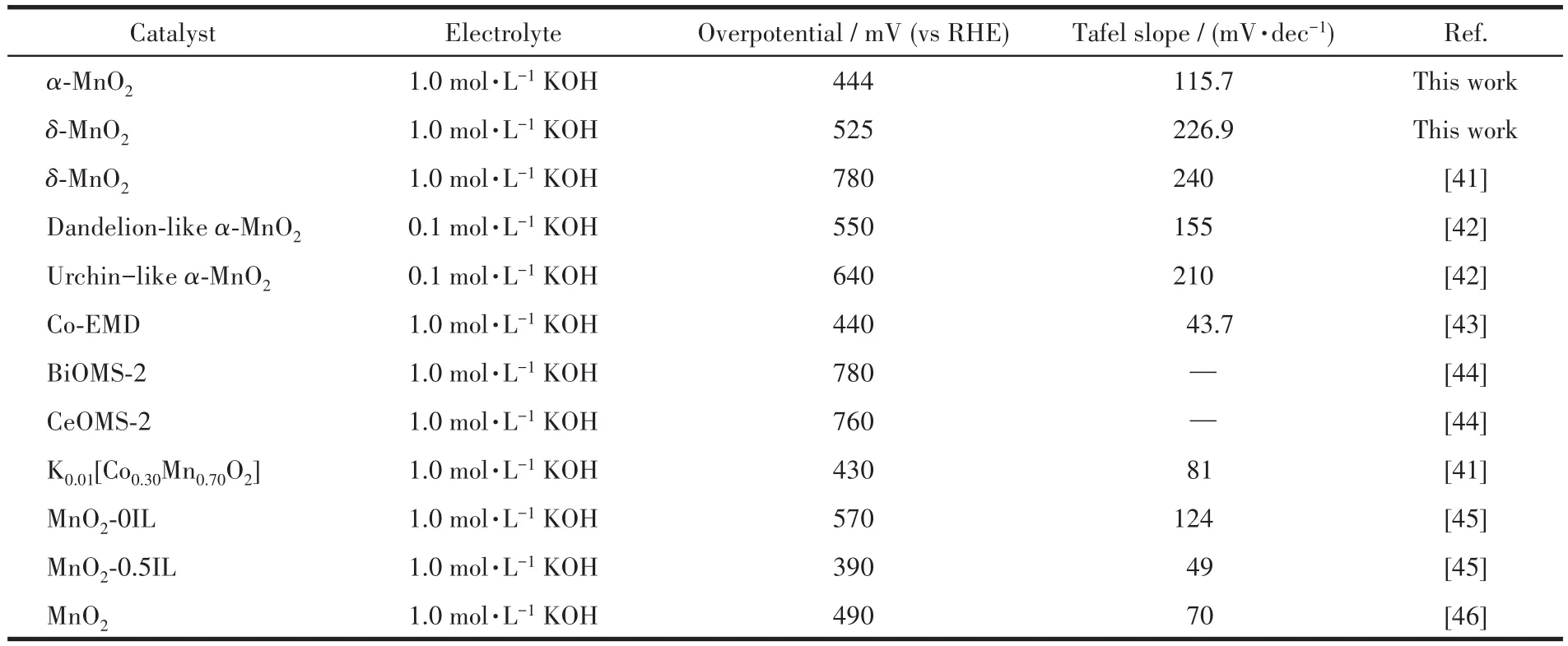
表1 不同MnO2催化剂的OER性能比较Table 1 Comparison of OER properties of the different MnO2 catalysts
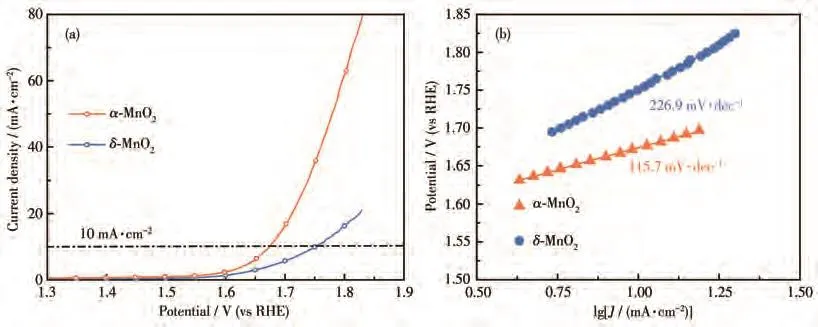
图4 α-MnO2和δ-MnO2在氧气饱和的1 mol·L−1KOH溶液、5 mV·s−1扫速下的(a)LSV曲线和(b)Tafel斜率曲线Fig.4 (a)LSV curves and(b)Tafel plots of α-MnO2 and δ-MnO2 in O2-saturated 1 mol·L−1KOH solutions at a scan rate of 5 mV·s−1
电化学活性面积(ECSA)是评价OER催化剂性能的另一个关键因素。ECSA的大小与双电层电容(Cdl)呈正相关关系[30]。在非法拉第区间1.13~1.34 V(vs RHE)记录样品在不同扫速下的CV曲线(图5a、5b),作电流密度差与扫速的关系图。通常来说,较大的ECSA意味着样品具备较好的电催化活性。如图5c所示,α-MnO2拥有的Cdl为27.14 mF·cm−2,略高于 δ-MnO2(22.33 mF·cm−2)。这可能是由于 α-MnO2具有大量的边缘共享di-μ-oxo桥活性位点,高的表面活性区为电解质和离子的扩散提供了具有活性位点的大的功能表面积和高的界面接触区[42]。
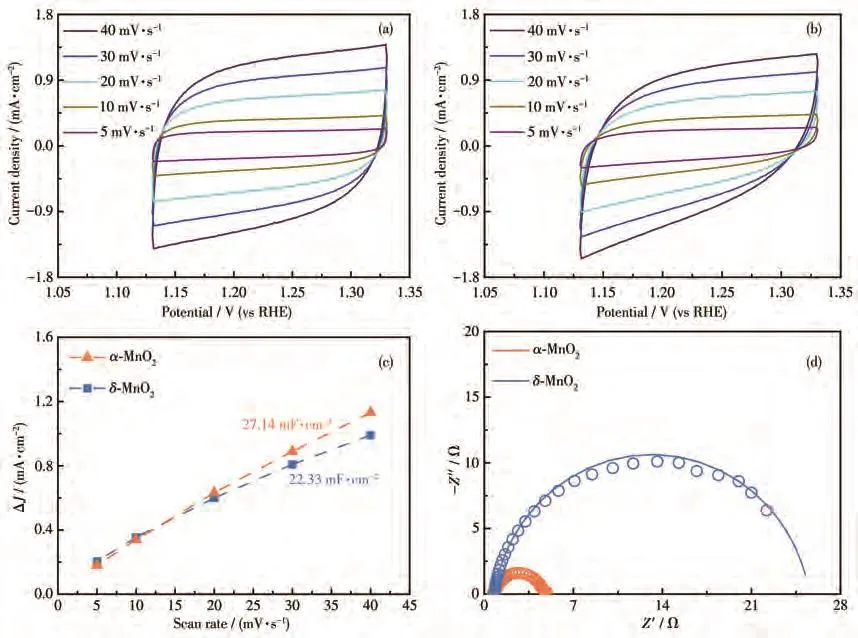
图5 (a)α-MnO2和(b)δ-MnO2在不同扫描速率下的CV图;α-MnO2和δ-MnO2的(c)双电层电容和(d)EIS谱图Fig.5 CV curves at different scan rates of(a)α-MnO2 and(b)δ-MnO2;(c)Double-layer capacitances and(d)EIS spectra of α-MnO2 and δ-MnO2
通过EIS测试来分析催化剂的电极动力学和阻抗大小。图5d为α-MnO2、δ-MnO2在偏压为0.55 V(vs Ag/AgCl)下测得的基于Nyquist图的EIS谱图,图中半圆的直径越大代表OER的反应阻抗越大。由EIS谱图可知,α-MnO2的半圆直径比δ-MnO2的小,α-MnO2的电荷转移阻力(Rct)为4.8 Ω,小于δ-MnO2的Rct(24.9 Ω),说明它拥有更快的电荷转移速率,更有利于OER反应的进行。
图6为催化剂在10 mA·cm−2的恒电流密度下电极电位随时间变化的计时电位曲线。在电解过程中的前30 min,δ-MnO2比α-MnO2电位的上升幅度更大,可能是由于δ-MnO2发生了严重的极化。经过12 h持续的电催化OER后,α-MnO2的电位只上升了约50 mV,而δ-MnO2在经过11 h的电解反应后,电位已上升至2.4 V,说明α-MnO2具有更好的稳定性。较低的反应温度导致δ-MnO2结晶性较差,在持续极化过程中,较弱的Mn—O键容易断裂[27],导致稳定性弱。反应温度的提高促进α-MnO2的热力学稳定性提高,表现出更优异的OER稳定性。
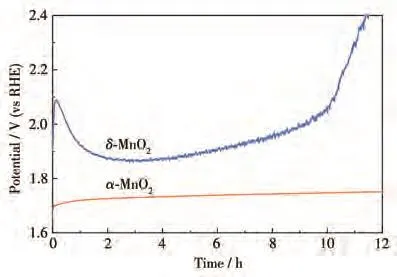
图6 α-MnO2和δ-MnO2在10 mA·cm−2电流密度下的稳定性测试Fig.6 Stability tests of α-MnO2 and δ-MnO2 under a current density of 10 mA·cm−2
2.4 性能分析
采用XPS分析了α-MnO2和δ-MnO2表面Mn和O元素的化学状态。图7a的全谱图显示α-MnO2和δ-MnO2均含有 Mn、O、Na和 K 元素。α-MnO2和δ-MnO2的Mn3sXPS谱图如图 7b所示,α-MnO2的Mn3s分裂能(ΔE)为 4.61 eV,大于δ-MnO2(4.42 eV)。Mn3s的多重态分裂能随Mn的平均氧化态(AOS)的增加而降低,根据Mn3s的ΔE,α-MnO2和δ-MnO2的AOS分别计算为3.77和3.97[47-48]。依据电中性原理,α-MnO2可能比δ-MnO2含有更多的表面氧空位和Mn3+[49]。图 7c为α-MnO2和δ-MnO2的 Mn2pXPS 谱图,642和654 eV处2个峰分别对应Mn2p3/2和Mn2p1/2的特征峰[50]。对Mn2p3/2特征峰进行分峰拟合得到641.9和643.1 eV两个峰,分别归属于Mn4+和Mn3+[49],α-MnO2表面的Mn3+和Mn4+的比例为3.45,高于δ-MnO2(2.09),说明α-MnO2表面具有更丰富的Mn3+。研究表明,在碱性介质中,Mn3+-OH通过质子耦合电子转移过程转化为Mn4+-OH,随后耦合2个相邻的末端氧原子释放O2[51-52]。催化剂表面富集的Mn3+可以提高对O−和OH−双重基团的吸附效率,从而促进Mn3+和Mn4+之间的氧化还原循环,有助于在OER中表现出优越特性[28,42,53]。从图7d的O1s谱图中可以发现2个峰:529.8 eV处的峰对应晶格氧(Olat),另一个531.3 eV的峰归属于表面吸附氧(Oads)。根据拟合峰结果计算得Oads和Olat的比例依次为α-MnO2(0.455)>δ-MnO2(0.387),这一顺序与OER催化活性一致,表明适当提高反应温度和添加浓硫酸能有效增强高表面活性氧的含量。氧分子通常吸附在氧空位上,表面吸附氧比晶格氧具有更高的表面迁移率,对OER活性有关键影响[54-55]。
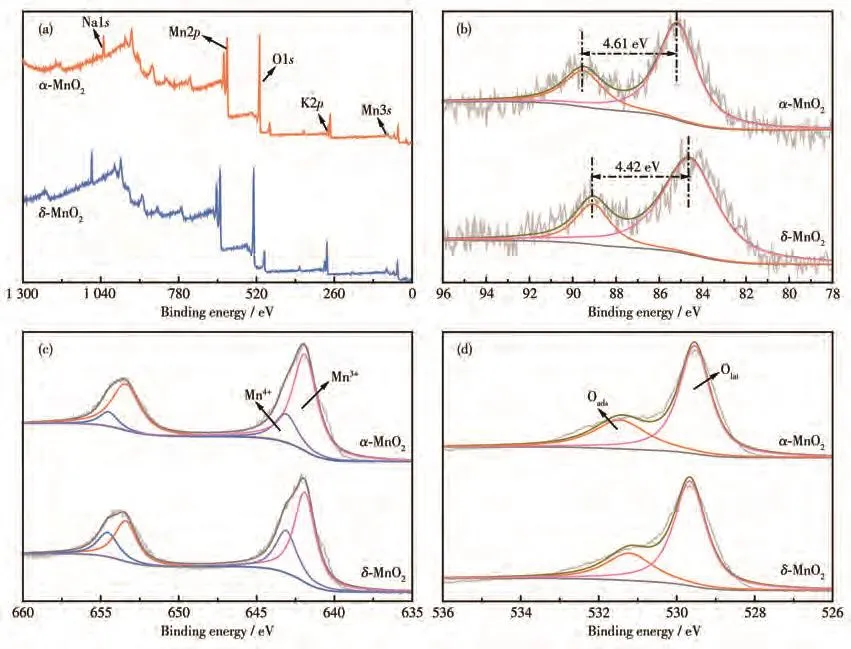
图7 α-MnO2和δ-MnO2的XPS图:(a)全谱图;(b)Mn3s、(c)Mn2p和(d)O1s高分辨谱图Fig.7 XPS spectra of α-MnO2 and δ-MnO2:(a)survey spectrum;high-resolution spectra of(b)Mn3s,(c)Mn2p,and(d)O1s
为了进一步分析α-MnO2的OER性能,在12 h稳定性测试后,对α-MnO2进行SEM、XRD和XPS表征,结果如图8所示。从图8a、8b可以看出,经过12 h稳定性测试后,α-MnO2仍然是纳米线阵列结构,其形貌与OER测试前的相似。如图8c所示,XRD峰信号与标准卡片(PDF No.44-0141)一一对应,说明α-MnO2电极结构稳定性较好。α-MnO2在稳定性测试前后的XPS谱图无明显差别,测试后Mn3s的ΔE为 4.69 eV,Mn2p中 Mn3+和 Mn4+的比例为 3.47,O1s中Oads与Olat的比例为0.454。这体现了α-MnO2的热力学稳定性可能是其维持良好OER稳定性的主要因素之一。
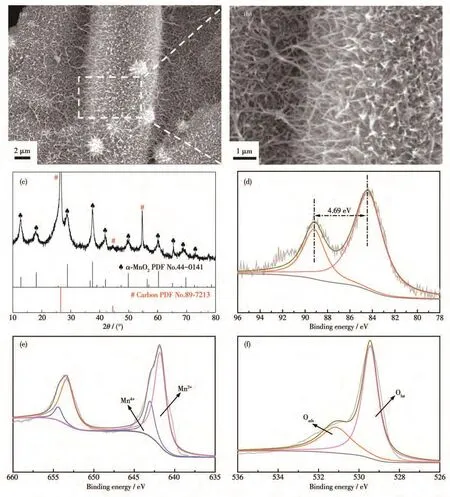
图8 稳定性测试后α-MnO2的(a、b)不同放大倍数SEM图、(c)XRD图及(d)Mn3s、(e)Mn2p、(f)O1s高分辨XPS谱图Fig.8 (a,b)SEM images with different magnifications,(c)XRD pattern,and high-resolution(d)Mn3s,(e)Mn2p,(f)O1s XPS spectra of α-MnO2 after stability tests
3 结论
综上所述,通过水热反应、改变反应温度和适当添加浓硫酸,成功合成α-MnO2纳米线和δ-MnO2纳米片阵列。结果表明,反应温度、添加浓硫酸对MnO2产物的晶相和形貌有显著的控制作用。排列整齐的α-MnO2纳米线阵列和δ-MnO2纳米片阵列能够提供良好的电子运输通道,直立的复合纳米阵列结构大大减小了气泡与电极之间的接触区域,有利于气泡的释放和离子的转移,增大了催化剂材料与电解液接触的面积,提高了OER活性和稳定性[16]。α-MnO2表面具有更高含量的Mn3+和氧空位,有利于OER过程中活性物种的快速转变,以及对OH−的吸附能力的提高。相比于δ-MnO2纳米片(10 mA·cm−2时过电位为 522 mV,Tafel斜率为 226.9 mV·dec−1),α-MnO2纳米线的OER活性显著增强,在10 mA·cm−2时过电位为444 mV,Tafel斜率115.7 mV·dec−1,反应温度的升高增强了MnO2的热力学稳定性,具有良好的长期耐久性。
猜你喜欢
杂志排行

无机化学学报的其它文章
- Recognition and Cell Imaging of Zn2+by Coumarin Derivative Fluorescence Sensor
- One⁃Pot Aqueous Synthesis and Cell Labeling Application of Glutathione Capped Cu⁃In⁃Zn⁃S Quantum Dots
- Preparation and Characterization of Copper Complexes of Schiff Base Ligands Synthesized In Situ from Spiropyran Derivative
- Syntheses and Luminescence of Three Zinc Complexes Constructed by Rigid 4⁃Substituted Bis(1,2,4⁃triazole)Ligand
- 三维花状Bi2WO6/BiOBr异质结的制备及对多种染料的降解性能
- 单分散SiO2纳米颗粒复合凝胶电解质的应用