高效液相色谱法检测乙酰半胱氨酸原料药中有关物质*
2022-02-17张承志谢育媛郭江红
张承志,谢育媛,郭江红
(湖北省药品监督检验研究院·国家药品监督管理局血液制品重点实验室·湖北省药品质量检测与控制工程技术研究中心,湖北 武汉 430064)
乙酰半胱氨酸又称N−乙酰基−L−半胱氨酸,作为一种经典化痰药物,常用于治疗呼吸道感染性疾病。研究发现,乙酰半胱氨酸有较强的抗氧化应激和促肺表面活性物质生成等作用,目前广泛用于呼吸系统、消化系统、心血管系统、泌尿系统疾病的防治[1−5]。药物中的杂质除了影响其纯度外,还可能影响药理活性或稳定性,造成药效偏差,甚至导致严重不良反应[6−7]。乙酰半胱氨酸是由盐酸半胱氨酸与醋酐的乙酰化反应生成的产物,原料药和生产工艺均可能产生L−胱氨酸、L−半胱氨酸、N,N′−二乙酰−L−胱氨酸、N,S−二乙酰−L−胱氨酸4种已知杂质。2020年版《中国药典(二部)》[8]收载的乙酰半胱氨酸中,尚未要求有关物质的检查;收载的乙酰半胱氨酸颗粒的有关物质采用高效液相色谱(HPLC)法控制单个未知杂质和总杂质的含量,未涵盖上述4种已知杂质。《英国药典2020版》(BP2020)[9]、《欧洲药典9.0版》(EP9.0)[10]、《日本药局方17版》(JP 17)[11]均收载了乙酰半胱氨酸原料药及其有关物质的检查项,《美国药典42版》(USP42)[12]仅收载了乙酰半胱氨酸原料药。本研究中参考EP9.0,建立了测定乙酰半胱氨酸原料药中4种已知杂质及其他未知杂质的HPLC法,并开展相关方法学验证,以提高乙酰半胱氨酸原料药的质量标准。现报道如下。
1 仪器与试药
1.1 仪器
Waters e2695/2489型高效液相色谱仪−紫外检测器(美国Waters公司);XP205型精密分析天平(瑞士梅特勒−托利多有限公司,精度0.01 mg);Milli−Q IQ7000型超纯水机(美国Millpore公司);LC−250型超声仪(济宁市中区鲁超仪器厂,功率250 W,频率33 kHz)。
1.2 试药
L−胱氨酸(杂质A)对照品(批号为LRAC3784,含量为99.9%),L−半胱氨酸(杂质B)对照品(批号为BCBZ1870,含量为99.1%),均购自德国Sigma−Aldrich公司;N,N′−二乙酰−L−胱氨酸(杂质C)对照品(批号为11.0,含量为100%),N,S−二乙酰−L−胱氨酸(杂质D)对照品(批号为7.0,含量为100%),均购自European Pharmacopoeia Reference Standard;乙酰半胱氨酸对照品(中国食品药品检定研究院,批号为140671−201903,含量为99.8%);乙酰半胱氨酸原料药(湖北新生源生物工程有限公司,批号分别为180701,180702,180703;武汉远大弘元股份有限公司,批号分别为202003003,202003004,202003005;浙江诚意药业股份有限公司,批号分别为0902−2020−03004,0902−2020−03005,0902−2020−03006);乙腈(色谱纯,德国默克公司);盐酸(分析纯,国药集团化学试剂有限公司);水为超纯水。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:ThermoODSHypersilC18柱(250mm×4.0mm,5µm);流动相:水(用磷酸调pH至3.0)−乙腈(97∶3,V/V);流速:1.0 mL/min;检测波长:220 nm;柱温:35℃;进样量:20 µL。混合对照品溶液在此色谱条件下的色谱图见图1。结果表明,4种已知杂质和乙酰半胱氨酸均能良好分离,各溶剂峰均无干扰。
2.2 溶液制备
取杂质A对照品21.09 mg,精密称定,置100 mL容量瓶中,加0.1 mol/L盐酸溶液10 mL超声使溶解,加水稀释并定容,摇匀,作为对照品溶液①;取杂质B对照品15.47 mg,精密称定,置100 mL容量瓶中,加0.1 mol/L盐酸溶液10 mL超声使溶解,加水稀释并定容,摇匀,作为对照品溶液②;取杂质C对照品10.51 mg,精密称定,置5 mL容量瓶中,加1 mol/L盐酸溶液0.5 mL超声使溶解,加水稀释并定容,摇匀,作为对照品溶液③;取杂质D对照品10.64 mg,精密称定,置50 mL容量瓶中,加0.1 mol/L盐酸溶液5 mL超声使溶解,加水稀释并定容,摇匀,作为对照品溶液④;分别精密量取对照品溶液①②③④各2,1,1,1 mL,置同一50 mL容量瓶中,加水稀释并定容,摇匀,即得对照品贮备液。取杂质A对照品、杂质B对照品、杂质C对照品、杂质D对照品、乙酰半胱氨酸对照品各10 mg,精密称定,置250 mL容量瓶中,加0.1 mol/L盐酸溶液25 mL超声使溶解,加水稀释并定容,摇匀,即得混合对照品溶液。取乙酰半胱氨酸原料药0.8 g,精密称定,置100 mL容量瓶中,加0.1 mol/L盐酸溶液10 mL超声使溶解,加水稀释并定容,摇匀,临用现配,即得供试品溶液①。将供试品溶液①于室温放置至少1 h,即得供试品溶液②。
2.3 方法学考察
线性关系考察:取2.2项下混合对照品溶液适量,作为对照品溶液①,取适量,加0.01 mol/L盐酸溶液准确稀释10倍,作为对照品溶液②。分别精密量取对照品溶液②20 µL,对照品溶液①10,20,40,60 µL,按2.1项下色谱条件进样测定,记录峰面积。以进样量(X,ng)为横坐标、峰面积(Y)为纵坐标进行线性回归,得杂质A,B,C,D的回归方程分别为YA=143.22XA+733.97(RA2=0.999 9),YB=76.09XB−2 991.43(RB2=0.999 4),YC=333.50XC−2 535.59(RC2=1.000 0),YD=764.74XD−26 888(RD2=0.999 7)。结果表明,杂质A,B,C,D的进样量分别在80.32~2 409.59 ng、81.58~2 447.38 ng、83.84~2 515.20 ng、80.64~2 419.20 ng
范围内与峰面积线性关系良好。
检测限与定量限确定:取2.2项下混合对照品溶液适量,用0.01 mol/L盐酸溶液逐步稀释,按2.1项下色谱条件进样测定。杂质A,B,C,D及乙酰半胱氨酸的检测限分别为5.04,8.64,3.70,5.69,8.34 ng,定量限分别为60.48,28.80,12.32,18.96,19.17 ng。
精密度试验:精密吸取2.2项下混合对照品溶液适量,按2.1项下色谱条件连续进样测定6次,记录峰面积。结果杂质A,B,C,D峰面积的RSD分别为0.15%,0.33%,0.15%,0.33%(n=6),表明仪器精密度良好。
重复性试验:取同一批(批号为202003003)样品0.8 g,精密称定,共6份,按2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积,按外标法计算各杂质的含量。结果杂质A,B,C,D含量的平均值分别为0.010%,0.004%,0.040%,0.004%,由于杂质含量较低,测得6份结果几乎无差异,表明方法重复性良好。
稳定性试验:取同一批(批号为202003003)样品,精密称定,置100 mL容量瓶中,加0.1 mol/L盐酸溶液10 mL使溶解,加水稀释并定容,摇匀,作为稳定性试验溶液,分别于0,1,3,5,7,9,11,13,15 h时按2.1项下色谱条件进样测定,记录峰面积。结果在15 h内,乙酰半胱氨酸和杂质A的峰面积均下降,杂质B,C,D的峰面积均增加,并出现1个保留时间约在3.6 min的降解杂质,表明供试品溶液极不稳定,需临用现配。
加样回收试验:取同一批(批号为202003003)样品0.4 g,精密称定,共6份,分别置100 mL容量瓶中,加0.1 mol/L盐酸溶液10 mL使溶解,精密加入对照品贮备液5 mL,加水稀释并定容,依法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积,并计算加样回收率。结果杂质A,B,C,D的平均加样回收率分别为101.03%,93.90%,98.23%,97.96%,RSD分 别 为5.21%,3.34%,4.91%,5.65%(n=6)。
破坏性试验:1)酸破坏。取同一批(批号为202003003)样品0.8 g,精密称定,置100 mL容量瓶中,加2 mol/L盐酸溶液5 mL,室温放置90 min,加2 mol/L氢氧化钠溶液调pH至中性,加0.01 mol/L盐酸溶液稀释并定容,按2.1项下色谱条件进样测定,记录色谱图。详见图2 A。2)碱破坏。取同一批(批号为202003003)样品0.8 g,精密称定,置100 mL容量瓶中,加2 mol/L氢氧化钠溶液5 mL,室温放置90 min,加2 mol/L盐酸溶液调pH至中性,加0.01 mol/L盐酸溶液稀释并定容,按2.1项下色谱条件进样测定,记录色谱图。详见图2 B。3)氧化破坏。取同一批(批号为202003003)样品0.8 g,精密称定,置100 mL容量瓶中,加5%过氧化氢溶液5 mL,室温放置90 min,加0.01 mol/L盐酸溶液稀释并定容,按2.1项下色谱条件进样测定,记录色谱图。详见图2 C。4)高温破坏。取同一批(批号为202003003)样品0.8 g,精密称定,置100 mL容量瓶中,沸水浴1 h,放冷,加0.01 mol/L盐酸溶液稀释并定容,摇匀,按2.1项下色谱条件进样测定,记录色谱图。详见图2 D。5)强光破坏。取同一批(批号为202003003)样品0.8 g,精密称定,置100 mL容量瓶中,2 600 lx照度下放置10 d,加0.1 mol/L盐酸溶液10 mL使溶解,加水稀释并定容,按2.1项下色谱条件进样测定,记录色谱图。详见图2 E。正常条件下,供试品溶液及空白溶剂色谱图见图2 F至图2 G。结果乙酰半胱氨酸在氧化破坏条件下极不稳定,在酸、碱破坏试验条件下,主峰峰面积下降明显,在高温、强光破坏试验条件下,主峰峰面积略有下降,表明该色谱条件能有效分离已知杂质峰及各相邻杂质峰,专属性良好。
2.4 样品含量测定
取9批样品各适量,分别按2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积,按外标法分别计算杂质A,B,C,D的含量。结果见表1。
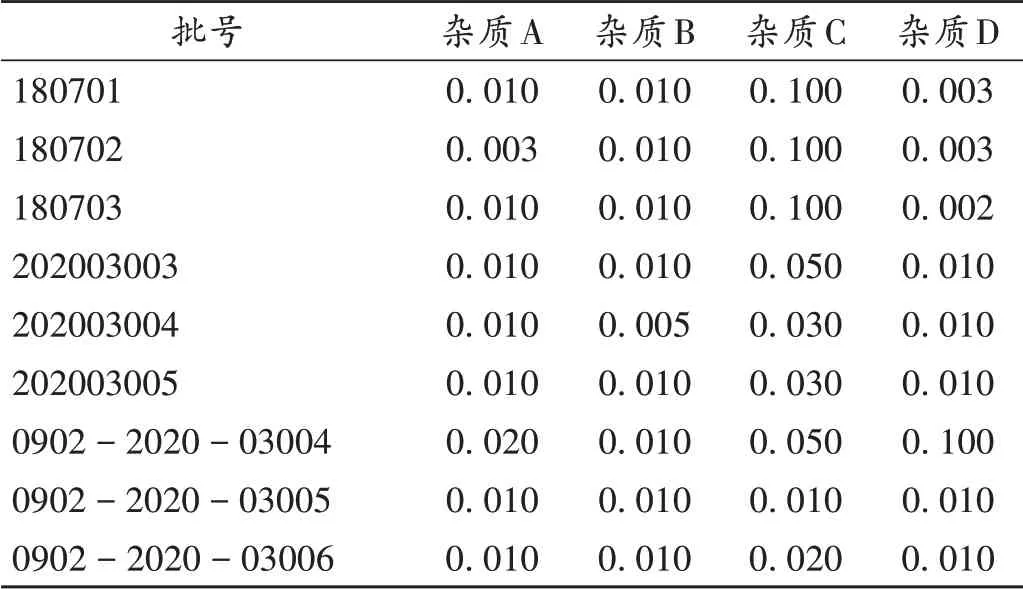
表1 乙酰半胱氨酸原料药中4种杂质含量测定结果(%)Tab.1 Results of content determination of four impurities in acetylcysteine APIs(%)
3 讨论
3.1 质量标准
EP10.3[13]与EP9.0收录色谱条件相同,在EP9.0的基础上严控了杂质限度,由各单个杂质不得过0.50%、总杂质不得过0.50%,改为不控制杂质A,杂质B,C,D依次不得过0.30%,0.20%,0.15%,单个未知杂质不得过0.10%,总杂质不得过0.50%。BP2020的标准同EP9.0。JP17采用HPLC法控制未知杂质和总杂质,依次不得过0.30%和0.60%。本研究中参考EP9.0,要求各单个杂质不得过0.50%,总杂质不得过0.50%,溶剂峰和灵敏度小于溶液主峰面积的单个杂质忽略不计。按拟订方法检测9批原料药,4种已知杂质含量均不超过0.10%,表明现行生产工艺较好。
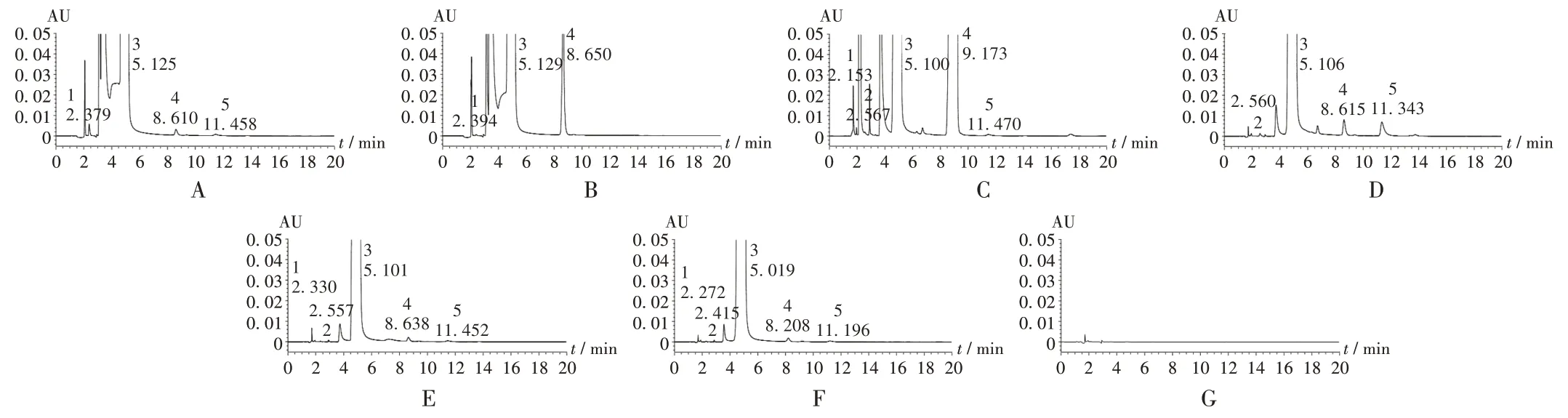
1.杂质A 2.杂质B 3.乙酰半胱氨酸4.杂质C 5.杂质DA−F.供试品溶液(条件分别为酸破坏、碱破坏、氧化破坏、高温破坏、强光破坏、正常条件)G.空白溶剂图2破坏性试验高效液相色谱图1.Impurity A 2.Impurity B 3.Acetylcysteine 4.Impurity C 5.Impurity DA−F.Test solution(conditions are acid destruction,alkali destruction,oxidation destruction,high−temperature destruction,strong−light destruction and normal conditions respectively)G.Blank solventFig.2 HPLC chromatograms of the destructive test
3.2 色谱条件优化
流动相pH选择:调节流动相A(水相)的pH分别为2.5,2.8,3.0,3.2,3.5,流动相水−乙腈(97∶3,V/V),其余色谱条件不变。结果显示:1)溶剂峰、杂质A峰与杂质B峰三者较难分离,随着pH的升高,溶剂峰与杂质A峰的分离度逐渐增加,当pH为3.0时,溶剂峰与杂质A峰才可分离;但随着pH的升高,杂质A峰与杂质B峰的分离度逐渐降低。2)主峰及4个杂质峰的对称性随pH的增加逐渐下降,主峰的理论板数从17 829(pH 3.0)降至3 308(pH 3.5)。综合考虑,最终选择流动相A为水(用磷酸调pH至3.0)。
流动相比例确定:设置流动相水(用磷酸调pH至3.0)−乙腈的比例分别为96∶4,97∶3,98∶2(V/V),结果表明,当有机相比例减少时,有利于杂质A与杂质B的分离[比例为98∶2(V/V)时,分离度为1.66],由于Hypersil ODS柱填料无法耐受高水相,故选择流动相比例为97∶3(V/V)。
色谱柱选择:由于杂质A极性较大,出峰较早,易包裹溶剂峰,故色谱柱需满足溶剂不干扰杂质A的测定,才能很好分离已知杂质峰和主峰。考察了Waters,Shiseido,GL,Thermo等品牌共10款C18柱[14],发现同时能满足以上要求的有Thermo ODS Hypersil C18柱和ODS−2 Hypersil C18柱,且前者分离效果更好,故被选用。
3.3 高效液相色谱仪选择
系统适用性试验中发现,同一系统适用性溶液在相同色谱条件下,杂质A与杂质B用戴安品牌高效液相色谱仪比Waters品牌的分离度更好,这可能与2种品牌仪器的管路体积、长度及色谱峰积分方式等因素有关。但考虑到杂质的检查方法应符合普遍适用性原则,较多企业使用Waters液相色谱仪,故选择Waters品牌。
3.4 溶液制备的优化
专属性试验考察中发现,强酸(2 mol/L盐酸溶液)会引起乙酰半胱氨酸主峰峰面积下降,故将EP9.0中供试品溶液配制“加1 mol/L盐酸溶液1 mL”改为“加0.1 mol/L盐酸溶液10 mL超声使溶解”。
3.5 待解决的问题
破坏性试验中发现,供试品溶液随放置时间的延长会产生相对保留时间(保留时间约3.6 min)约0.7的杂质,该杂质的峰面积随放置时间的延长而增大。EP9.0要求供试品溶液①临用现配,供试品溶液①至少放置1 h后产生的保留时间约为3.3 min的杂质峰(2−甲基−2−噻唑啉−4−羧酸峰)可忽略不计。JP17也要求供试品溶液临用现配。考虑是样品溶于盐酸溶液后不稳定导致降解杂质产生,并非样品本身的降解杂质,故未将降解杂质纳入限度考虑,并忽略不计。由于时间仓促,本研究中未进一步研究相对保留时间约0.7的降解杂质。
3.6 方法评价
本方法灵敏度高,准确度和重复性均较好,可用于检测乙酰半胱氨酸原料药中有关物质。
