CORRECT介导的人HBB-28基因定点突变细胞株的建立及其应用*
2022-02-09刘永祥麦庆云黎允诗梅珺琰梁爱军彭新良周瑞鸿周少虎
刘永祥,麦庆云,黎允诗,梅珺琰,梁爱军,彭新良,周瑞鸿,周少虎**
(1.广州中医药大学第一附属医院生殖医学科,广东广州 510405;2.中山大学附属第一医院生殖医学中心,广东广州 510080;3.广州医药研究总院有限公司,广东广州 510220)
β-地中海贫血(简称β-地贫)是由人β-珠蛋白(HBB)基因突变引起的,是我国南方发病率最高的单基因遗传病[1]。其中,HBB-28(A>G)突变是我国β-地贫患者携带的5种最常见的HBB突变之一[2]。与其他亚型不同,HBB-28(A>G)突变位于起始密码子上游28个碱基的启动子区内,突变的产生破坏了转录因子ATA box的结合,降低了HBB的RNA表达[3]。纯合子或复合杂合子-28(A>G)突变的患者可出现重度贫血或中间型贫血[4],然而,由于纯合子患者临床材料有限,并且其基因突变位于非编码区,目前非编码区突变影响相关基因转录组表达水平的研究较少。
近年来,由于操作简单、成本低和效率高等优点,规律间隔成簇短回文重复序列(Clustered Regularly Interspaced Short Palindromic Repeats,CRISPR)技术被广泛应用于各种生物体的基因组编辑[5]。利用CRISPR/Cas9基因编辑技术在特定位点制造特定的DNA双链断裂(DNA Double Strand Breaks,DSBs)和同源定向修复(Homology Directed Repair,HDR)以引入或纠正特定的基因突变已经成为首选的方法[6]。本研究在CRISPR/Cas9技术的基础上,通过沉默突变阻断连续的再次靶向或再次切割的CRISPR/Cas靶点(Consecutive re-Guide or re-Cas steps to Erase CRISPR/Cas-blocked Targets,CORRECT)的新型无痕基因组编辑技术[7],在人HBB基因上引入HBB-28(A>G)点突变,高效建立人β-地贫HBB-28(A>G)HEK293T纯合突变细胞株,为后续疾病的细胞和分子基础研究,以及建立基因突变修复方法奠定基础。
1 材料与方法
1.1 材料
1.1.1 细胞株和质粒
HEK293T细胞由本实验室保存,CRISPR/Cas9质粒PX459购自美国Addgene公司,pCMV-GFP-p62购自上海碧云天生物技术有限公司。
1.1.2 主要试剂和仪器
限制性核酸内切酶BbsⅠ、T7EⅠ、AatⅡ、T4连接酶、T4磷酸化酶均购于美国NEB公司,小向导RNA (small guide RNA,sgRNA)以及PCR引物均由生工生物工程(上海)股份有限公司合成,单链寡核苷酸(single-stranded Oligo DNA Nucleotides,ssODNs)由苏州金唯智生物科技有限公司合成,Trans 5α感受态细胞购自北京全式金生物技术有限公司,质粒提取试剂盒购自天根生化科技(北京)有限公司,X-tremeGENE HP DNA细胞转染试剂盒购于上海罗氏制药有限公司。荧光倒置显微镜为德国徕卡(Leica)仪器有限公司产品,PCR基因扩增仪为赛默飞世尔科技(中国)有限公司产品。
1.2 方法
1.2.1 sgRNA的设计与CRISPR/Cas9表达质粒构建
查询人HBB基因序列(Genbank:NC_000011.10;https://www.ncbi.nlm.nih.gov/),利用网站https://crispr.mit.edu设计sgRNA靶点。根据确定的sgRNA靶点序列合成2对互补引物,sgRNA均为正义链序列靶点,因此,需在引物5′端加上BbsⅠ酶的黏性末端,其中,CACC加在正向引物前,AAAC加在反向引物前(表1)。sgRNA引物磷酸化与退火:分别取1 μL T4磷酸化酶、1 μL正向引物和1 μL 反向引物(浓度均为100 μmol),混匀后进行5′端磷酸化修饰,并经退火形成双链(37℃,30 min;95℃,10 min)。双链sgRNA与线性化CRISPR/Cas9质粒连接:分别取1 μL T4连接酶、2 μL退火产物,以及100 ng经BbsⅠ酶处理的线性化二合一CRISPR/Cas9质粒,混匀后于16℃连接过夜,获得PX459-sgRNA1-Cas9和PX459-sgRNA2-Cas9的共表达质粒。转化(热激法):按照Trans 5α感受态细胞的产品说明书,将10 μL连接产物转化入100 μL感受态细胞中,轻轻摇匀,冰上放置30 min,42℃水浴中热激45 s,迅速置于冰上冷却2 min,涂布于含50 μg/mL氨苄青霉素(Amp)的LB固体平板,37℃倒置培养16-18 h。测序验证:分别挑取8个单克隆菌落至LB液体培养基中,37℃、220 r/min摇床扩增培养12-16 h,测序鉴定并挑选连接成功的PX459-sgRNA1-Cas9和PX459-sgRNA2-Cas9共表达阳性克隆,根据天根质粒小量提取试剂盒说明书提取阳性克隆质粒。

表1 sgRNA互补引物序列
1.2.2 ssODNs序列设计
选择突变位点左右两侧各50 bp序列作为同源臂,合成外源性同源重组模板ssODNs (图1)。设计ssODNs时采用CORRECT技术[7],即在不引起氨基酸移码突变或错义突变的前提下,为防止基因组DNA成功突变后被CRISPR/Cas9系统二次切割,依据氨基酸的简并性,在前间隔序列的邻近基序(Protospacer Adjacent Motif,PAM)附近引入同义突变碱基(A>C),重组后以终止Cas9 蛋白切割活性。因为HBB-28(A>G)位于非编码区,故本研究均用阻断突变(Blocking Mutation Base)表示。为方便后续的鉴定,在引入阻断突变碱基的同时,引入一个新的限制性内切酶AatⅡ识别位点(5′-GACGTC-3′)。如图1所示,ssODN1序列为正向序列,ssODN2为反向序列。
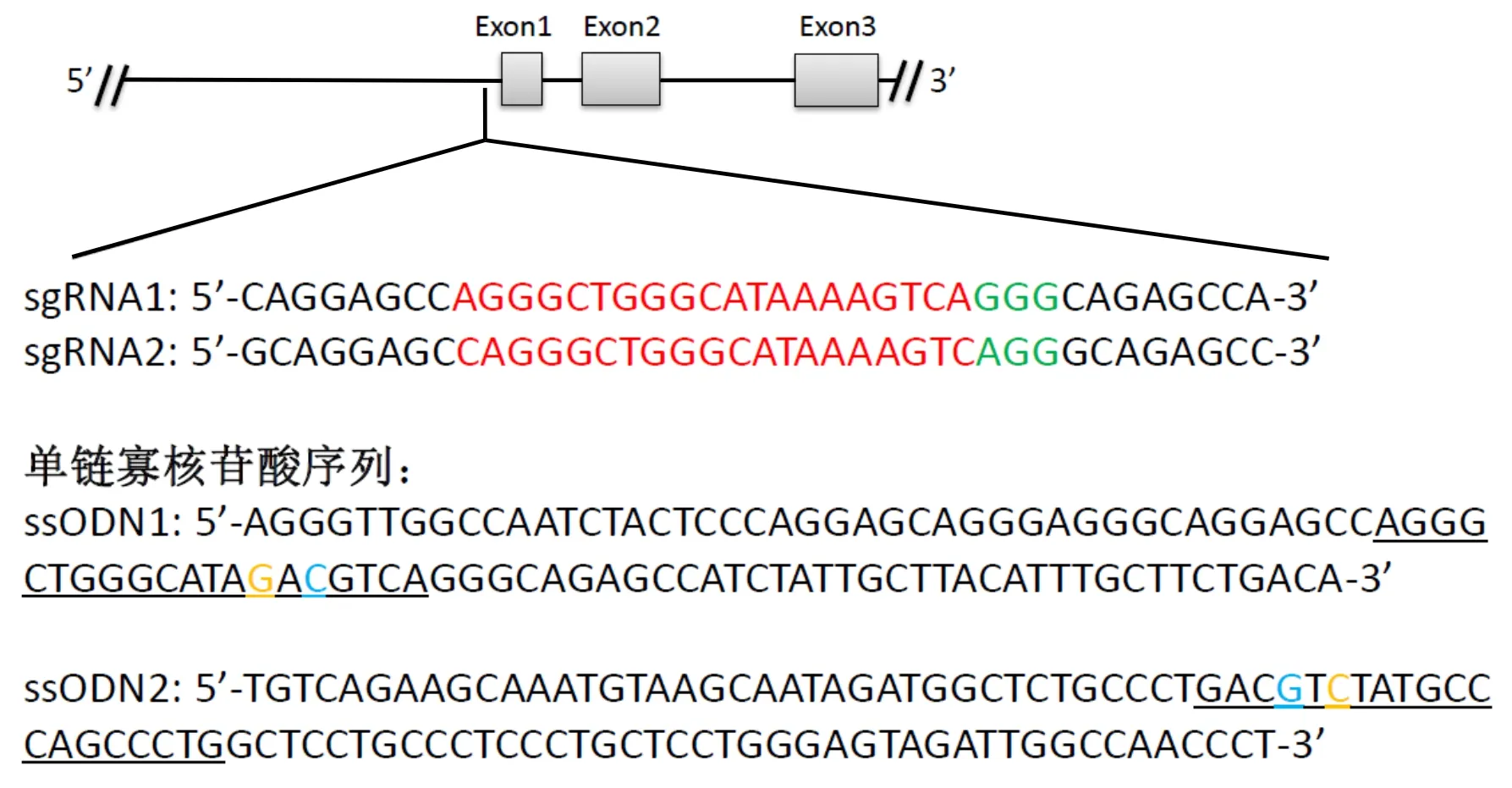
The red font is sgRNA sequence,the green font is PAM sequence,the yellow font is HBB-28 (A>G) mutation site,the blue font is A>C blocking mutation site,the lower horizontal line is the target sequence
1.2.3 脂质体转染
转染前于细胞培养瓶中复苏HEK293T细胞,使用含有10%的新生牛血清(FBS)、1%双抗(100 U/mL青霉素和链霉素)的DMEM培养基培养,当细胞传代2-3次性状稳定且状态较好时,将细胞消化后铺于细胞六孔板中,待六孔板中每孔的HEK293T细胞密度汇合到该孔总细胞的70%-80%时,使用X-tremeGENE HP DNA细胞转染试剂盒进行转染操作。在HEK293T细胞中脂质体分别转染1 μg、2 μg和3 μg含绿色荧光蛋白(Green Fluorescent Protein,GFP)的pCMV-GFP-p62表达载体,48 h后在荧光显微镜下观察GFP的表达丰度,评估转染效率,选择合适的质粒浓度进行转染。
转染:将脂质体X-tremeGENE HP、PX459-sgRNA1-Cas9/PX459-sgRNA2-Cas9共表达质粒、ssODN1/ssODN2和无血清培养基室温平衡至15-25℃,短暂漩涡混匀脂质体X-tremeGENE HP;用无血清培养基将PX459-sgRNA1-Cas9或PX459-sgRNA2-Cas9共表达质粒稀释至终浓度为1 μg质粒DNA/100 μL无血清培养基(0.01 μg/μL),轻柔混匀;再加入1.5倍PX459-sgRNA1-Cas9或PX459-sgRNA2-Cas9共表达质粒体积的脂质体(脂质体切勿接触到塑料管壁),室温孵育15-20 min,得到转染复合物;用吸管沿着边缘轻轻吸去六孔板中的细胞培养液,再加入2 mL新鲜的含有10%的FBS、1%双抗的DMEM培养基后,将处理好的转染复合物逐滴全部加入,轻轻摇匀,培养24 h后弃去培养液,并更换为2 mL含有0.6 μg/mL嘌呤霉素(Puromycin)的无血清培养基进行药物筛选,继续培养48 h,收集孔内的一半细胞并提取基因组DNA,PCR扩增含有突变位点的片段进行测序鉴定,得到作用sgRNA1和sgRNA2靶点的阳性克隆。
1.2.4T7EⅠ实验与整合效率检测
如图1所示,在ssODN发生整合后,会在1号外显子上游28个碱基处引入HBB-28(A>G)点突变和1号外显子上游26个碱基处引入阻断突变碱基(A>C),此时将得到一个新的限制性内切酶AatⅡ识别位点(5′-GACGTC-3′)。为了验证不同靶点的切割效率,以及正、反链ssODN的整合效率,将实验分为4组:①sgRNA1+ssODN1、②sgRNA1+ssODN2、③sgRNA2+ssODN1、④sgRNA2+ssODN2。采用1.2.3节的方法将各组进行脂质体转染48 h后收取HEK293T细胞,收集野生型293T细胞和实验组①-④部分细胞提取基因组DNA,PCR扩增含靶点的片段,引物为HBB-FP:5′-GCAATTTGTACTGATGGTATGG-3′;HBB-RP:5′-ATAACAGCATCAGGAGTGGAC-3′。PCR 扩增反应条件为预变性94℃、4 min;变性94℃、30 s,退火56℃、30 s,延伸72℃、30 s,35个循环;总延伸72℃、10 min,4℃保存。最后分别取200 ng PCR产物进行T7EⅠ(T7EⅠ 0.5 μL和相应buffer 1 μL,共10 μL体系,37℃,0.5 h)和AatⅡ(AatⅡ 0.5 μL和相应buffer 1 μL,共10 μL体系,37℃,1 h)酶切,使用10×Loading buffer终止酶切反应后,取8 μL酶切产物进行1%琼脂糖凝胶电泳,选择有切割效率的PCR产物进行Sanger测序。
凝胶成像后用ImageJ软件进行灰度分析,比较各组切割效率和整合效率。切割效率用T7EⅠ酶切活性表示,整合效率用AatⅡ酶切活性表示。切割效率[8]=1-[c/(a+b+c)]0.5(其中a、b分别为凝胶图中被切割2条电泳条带对应ImageJ软件的峰面积;c为未被切割条带的峰面积,也是最大的峰面积)。根据被整合细胞占T7EⅠ切割阳性细胞的百分比,设定相对整合效率的计算公式:相对整合效率=(AatⅡ酶切活性/T7EⅠ酶切活性)×100%。被AatⅡ切割越多,说明该组发生基因重组或整合的细胞越多,整合效率越高。
1.2.5 单克隆培养与鉴定
根据酶切实验的结果,选择整合效率最高的实验组采用96孔板计数稀释接种的方法[9]进行单克隆筛选,细胞采用含有10%的FBS、1%双抗的DMEM培养基,于37℃、5% CO2培养箱培养4-5 d后,观察除去双细胞和多细胞孔,对细胞形态正常且单一细胞孔做标记;继续培养7-9 d,待细胞长满整个孔后,吸取孔内2/3的单克隆集落细胞提取基因组DNA,挑选10个单克隆集落细胞,采用1.2.4节的PCR方法扩增含有突变位点的片段,并进行测序鉴定,获得含目标纯合突变的单克隆细胞株。
2 结果与分析
2.1 CRISPR/Cas9表达质粒构建
根据图2的质粒测序分析结果可知,PX459-sgRNA1-Cas9和PX459-sgRNA2-Cas9共表达质粒构建成功。
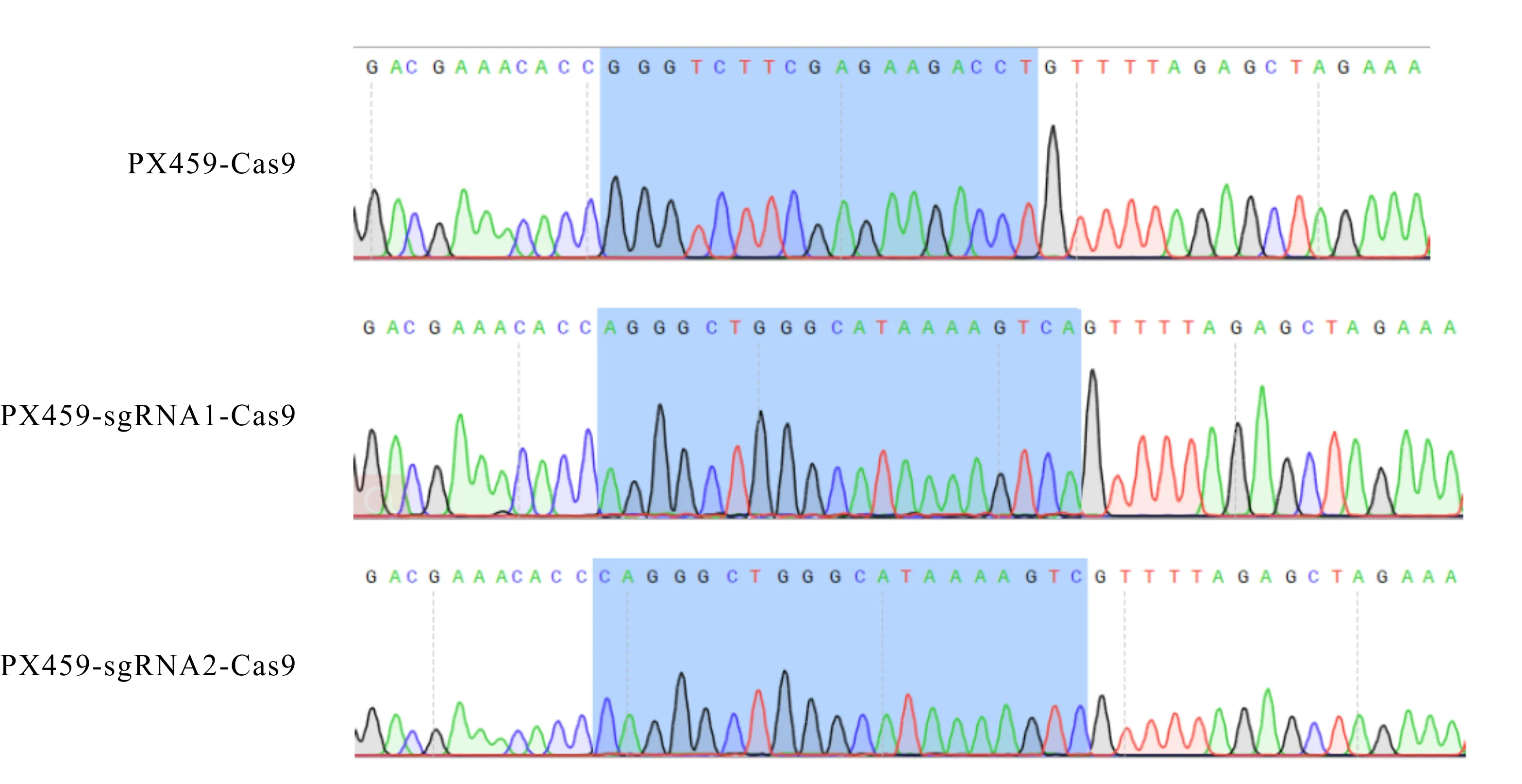
图2 PX459-sgRNA1-Cas9和PX459-sgRNA2-Cas9共表达质粒测序结果
2.2 脂质体转染效率
荧光显微镜下观察发现,转染1 μg质粒时不到20%的细胞表达GFP,转染2 μg质粒时表达GFP的细胞比例达到80%,转染3 μg质粒时表达GFP的细胞比例在85%以上(图3)。为了减少转染的毒性同时又达到较好的转染效果,选择转染2 μg质粒进行后续实验。
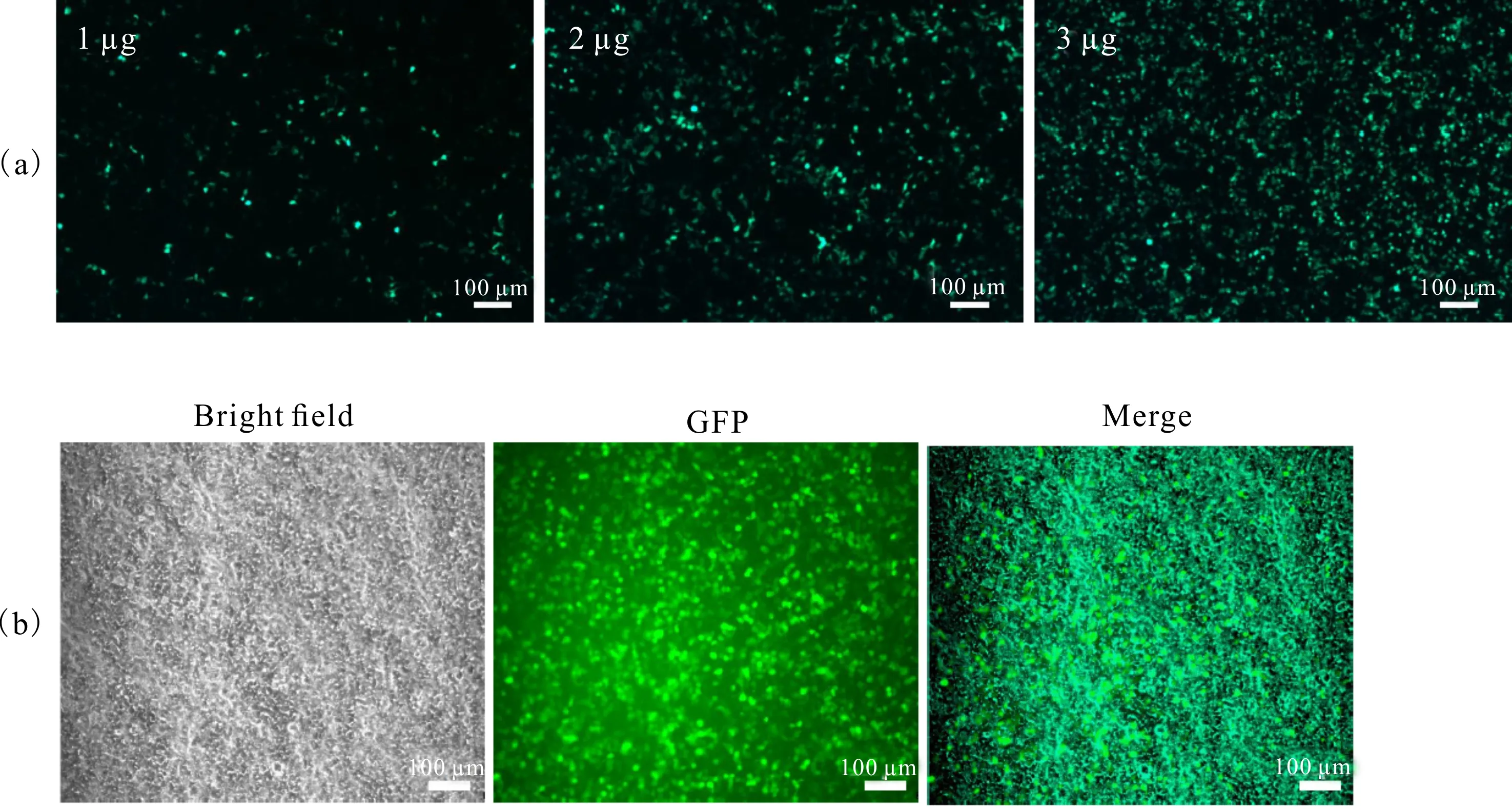
(a) Fluorescence spectra of transfected by 1 μg,2 μg and 3 μg GFP plasmids;(b) The bright field and green fluorescence image of 2 μg GFP plasmid transfection
2.3 HBB-28基因(A>G)定点突变HEK293T细胞株的建立
2.3.1 sgRNA切割效率和ssODNs整合效率验证
sgRNA切割效率实验(T7EⅠ酶切活性):PCR扩增包含有突变位点在内的片段,实验组①-④的PCR产物总长度是578 bp,且均表现出切割活性,酶切后分为3条条带:原有条带578 bp,以及被切割后的372 bp和206 bp两条条带(图4)。结合灰度分析,实验组①-④的切割效率分别为27.19%、22.72%、18.62%和21.49%(表2)。对于HBB-28基因,活性最高的是sgRNA1,因此,后续的基因定点突变实验选用sgRNA1进行。
ssODNs整合效率实验(AatⅡ酶切活性):当ssODNs整合至目标位置时,会被AatⅡ酶识别并切割,如图4的电泳图及表2的灰度分析结果所示,实验组①-④均有AatⅡ酶的酶切活性,整合效率分别为26.41%、22.25%、14.78%和12.84%,说明这4组均有ssODNs整合。
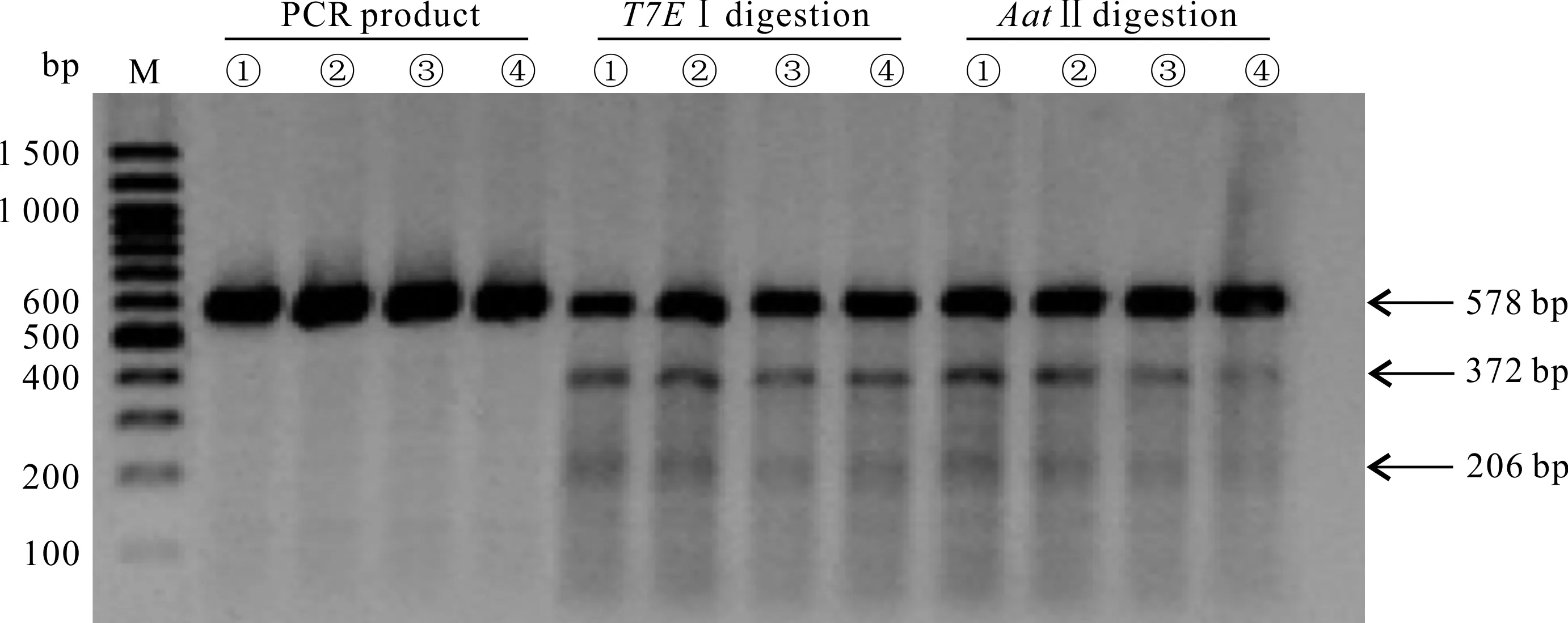
M:Marker;①:sgRNA1+ssODN1;②:sgRNA1+ssODN2;③:sgRNA2+ssODN1;④:sgRNA2+ssODN2
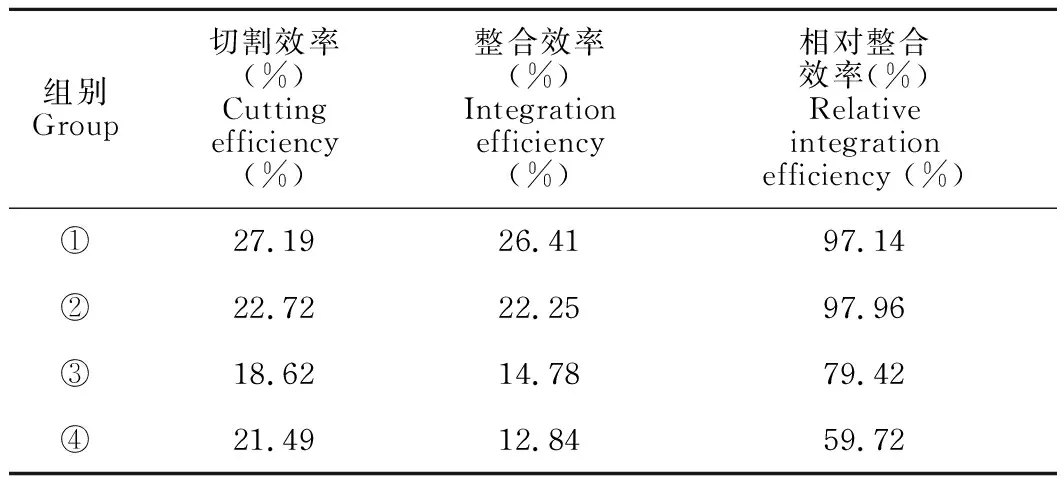
表2 各组切割效率、整合效率和相对整合效率
根据表2汇总的各组切割效率、整合效率和相对整合效率可知,实验组①的切割效率和整合效率最高,其相对整合效率为97.14%,与相对整合效率最高的实验组② (97.96%)相差不大。为了减少实验结果的误差,对实验中的PCR产物进行T7EⅠ酶切和AatⅡ酶切的重复实验,结果与初次实验差异不大。因此,选择实验组①进行后续的单克隆筛选实验。
2.3.2 测序鉴定
结合图4和表2的灰度分析,以及图5峰值结果,实验组①-④均发生了切割和整合,引入了β-地贫基因HBB-28(A>G)和阻断突变碱基(A>C),且实验组①和②的杂合峰值比实验组③和④高(图5),说明实验组①和②的切割效率和整合效率高,但未进行单克隆挑选,故显示为杂合的峰图。
2.4 β-地贫基因HBB-28(A>G)定点突变的单克隆HEK293T细胞株的建立
如图6所示,单克隆HEK2937细胞株同时引入了HBB-28(A>G)和阻断突变(A>C)碱基。10个单克隆中还包含5个杂合突变、1个非目标突变纯合细胞株以及3个无相应碱基突变的野生型细胞株。

The blue arrow shows HBB-28 pathogenic point mutation base (A>G),and the red arrow shows blocking mutation base(A>C)
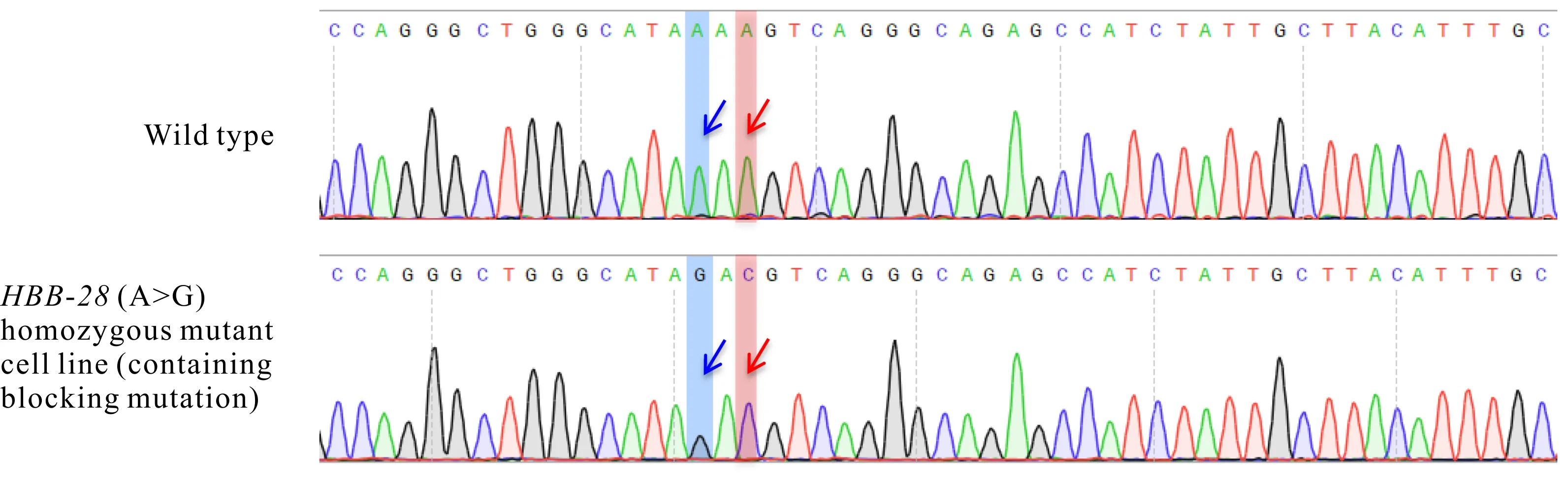
The blue arrow shows HBB-28 pathogenic point mutation base (A>G),and the red arrow shows blocking mutation base(A>C)
3 讨论
CRISPR/Cas9基因编辑技术是继锌指核酸酶(Zinc-Finger Nucleases,ZFNs)和转录激活因子样效应物核酸酶(Transcription Activator-Like Effector Nucleases,TALENs)技术之后出现的第三代新型基因编辑技术。CRISPR/Cas9基因编辑技术通过导向sgRNA招募Cas9蛋白在细胞内靶基因特定位点进行特异切割形成DNA双链断裂,进而诱发同源重组和非同源末端连接的自我修复过程,实现对基因组特定靶点的编辑[5,10]。该技术操作便捷、重复性好,而且易于设计、插入效率高,已在医学、药物、生命科学等多领域得到广泛应用[11]。
在哺乳动物细胞中,基因HDR频率相对少见,大多数情况下DSBs由非同源连接(Non-Homologous End Joining,NHEJ)修复[12]。然而,HDR介导的基因插入在人类疾病模型制备、基因治疗和农业遗传改良等方面具有重要作用,但是其效率很低[13]。因此,如何提高HDR的频率对提高编辑效率显得格外重要。其中,单链供体ssODNs模板的应用,有助于提高HDR的频率。与利用双链DNA(double strand DNA,dsDNA)模板或其他遗传操作进行小片段插入、缺失或点突变不同,对于短片段的插入,ssODNs作为供体模板具有更高的基因插入效率[14]。另外, dsDNA同源模板受拓扑结构、同源臂长度等影响,构建过程需要数周,而ssODNs合成时间短,且不需要任何筛选标记就可以一步完成基因精确修复,不会随机整合到基因组中,是一种更加安全、精确的基因编辑方法[15,16]。
Kwart等[7]和Paquet等[17]利用CRISPR/Cas9和ssODN在人类多功能干细胞中引入与疾病相关的致病突变时发现,高达95%的已编辑位点的序列会被再次编辑,造成额外的indel突变破坏。利用CORRECT技术能够有效阻断多个位点的再次编辑,这些阻断突变碱基能以高效率、高精度选择性引入单等位或双等位基因序列,使HDR的编辑精度提高到每个等位基因的10倍,阻断突变的位点越接近PAM位点,HDR的效率越高[7,17]。为了提高HDR的频率,本研究一方面利用CORRECT基因编辑新技术在CRISPR/Cas9靶向所需PAM序列附近插入同义突变碱基到HDR模板中,在很大程度上阻止不良的再次编辑,提高了ssODNs的相对整合效率(达97.14%),获得了纯合的单克隆HBB-28(A>G)细胞株。另一方面,本研究在PAM前5个碱基处引入限制性内切酶AatⅡ识别位点,同样得到较好的编辑和整合效率。比较ssODNs的正反方向对整合效率的影响发现,sgRNA1和sgRNA2均为正义链序列的靶点,对于编辑效率高的sgRNA1靶点,正、反向ssODNs的整合效率分别为26.41%和22.25%,相对整合效率分别为97.14%和97.96%,表明正、反方向的ssODNs对相对整合效率影响不大。对于编辑效率相对较弱的sgRNA2靶点,正、反向ssODNs的整合效率分别为14.78%和12.84%,相对整合效率分别为79.42%和59.72%,表明对sgRNA2靶点而言,正向ssODN1的相对整合效率更高。但该结果是否适用于其他靶点,需要进一步验证。尽管针对单个等位基因的阻断突变碱基(同义突变)的引入不影响氨基酸序列,但是引入的突变碱基是否在DNA或RNA水平影响细胞的转录调控或内部功能尚不清楚。遗传密码被破解以来,人们通常认为遗传密码具有简并性,同义突变是无害的,不会改变蛋白质序列[18,19]。然而Shen等[20]最近的研究发现大多数同义突变是非常有害的,他们通过改造酿酒酵母基因组中21个有代表性的基因,制造了8 341个突变体,结果显示至少75%的同义突变显著损害酿酒酵母的适应度,且损害幅度超过0.1%,是自然选择在酿酒酵母中所能感知的最小适应度变化的10 000倍。但是该实验是在单倍体芽殖酵母中进行的,后期需要在不同生物体及其杂合状态下做进一步的实验验证。
本研究虽然未进行基因点突变后的细胞功能验证,但是在方法学上提供了一种新型、高效的点突变编辑方法,且该方法同样适用于特定基因突变位点的修复,为后续的研究奠定了模型和方法基础。
