重组Pol D DNA聚合酶的纯化及其功能初步研究*
2022-02-09张雪松李德凤薛姜珊陈孜孟蒙健宗
张雪松,李德凤,薛姜珊,陈孜孟,李 卓,蒙健宗
(1.广西大学生命科学与技术学院,广西南宁 530005;2.自然资源部第三海洋研究所,福建厦门 361000;3.广西农业职业技术大学食品药品研究院,广西南宁 530007)
DNA复制在生命领域中是必不可少的生物学过程,DNA聚合酶在DNA复制过程中起着十分关键的作用,它确保了遗传信息的准确复制和传递。迄今报道的DNA聚合酶家族有7个,根据对其氨基酸序列和结构的分析可划分为A、B、C、D、E、X和Y家族[1-7],其中D家族(Pol D)DNA聚合酶是在古菌中被发现的,并且只存在于古菌中[5]。1997年Uemori等[8]从超嗜热古菌Pyrococcusfuriosus中克隆出Pol D DNA聚合酶基因,该聚合酶由DP1和DP2两个亚基组成,大小分别为69 294 Da和143 161 Da,且两亚基需要共表达或混合存在时才会各自表现出较强的活性;1998年Ishino等[5]从古菌Methanococcusjannaschii中克隆表达了与P.furiosusDNA聚合酶同源的聚合酶蛋白,并发现其DP1亚基有3′→5′外切核酸酶活性;同时发现,相比于双链DNA,DP1更容易降解单链DNA。研究发现大部分古菌中存在B家族(Pol B)与Pol D两种DNA聚合酶,若将细胞内的Pol B DNA聚合酶基因敲除,古菌依旧可以存活;若将细胞内的Pol D DNA聚合酶基因敲除,则古菌无法存活[9]。由此可见Pol D DNA聚合酶在古菌DNA复制过程中的重要性。相比于Pol A、Pol B等家族的DNA聚合酶,国内外对Pol D DNA聚合酶的研究仍然较少,多种古菌中Pol D DNA聚合酶的理化性质和功能仍处于未知状态。
我国自然资源部第三海洋研究所从采集的海底热液口样品中分离得到一株超嗜热古菌Thermococcussp.4557 (CGMCC 1.517 2),该菌是一种严格厌氧的超嗜热硫还原古菌[10],前期已克隆表达其小亚基DP1和大亚基DP2,并研究了DP1与Sld5和Psf1蛋白复合体(Sld5 and Psf1 Complex,GINS 51)、GINS关联核酸酶(GINS-Associated Nuclease,GAN)等相关蛋白的体外相互作用[11],但还未研究该Pol D DNA聚合酶大、小两个亚基的功能及其相互作用,以及GINS 51蛋白对小亚基外切酶活性的影响等。本研究分别纯化重组表达Thermococcussp.4557 Pol D DNA聚合酶小亚基DP1和大亚基DP2,并利用荧光标记核酸对其体外功能进行研究。
1 材料与方法
1.1 材料
1.1.1 表达质粒与重组蛋白
分别用于表达超嗜热硫还原古菌Thermococcussp.4557 Pol D DNA聚合酶小亚基DP1和大亚基DP2的表达质粒pET21a/His-PolD-(S)、pET21a/His-PolD-(L)[11],以及纯化的重组表达Thermococcussp.4557 GINS 51蛋白,均由自然资源部第三海洋研究所提供。
1.1.2 试剂
Ni-NTA树脂、Q阴离子层析树脂、DEAE树脂和Butyl树脂购自GE Healthcare Life Sciences,牛血清白蛋白(BSA)购自上海阿拉丁生化科技股份有限公司,dNTP购自New England Biolabs,Trition X-100购自Takara Bio Group。
单链DNA模板ZXS001序列:5′-GTAACGC-
CAGGGTTTTCCCAGTCACGACGTTGTAAAA-
CGACGGCCAGTGCCAAGCTTGCATGCCTGC-
AGGTCGACTCTAGAGGATCCCCGGGTACCG-
AGC-3′。
荧光引物Cy3-ZXS002序列:5′-GCTCGGTAC-
CCGGGGATCCTCTAGAGTCGA-3′。
荧光底物Cy5-0A01序列:5′-GGGGCGAGTCCAGGTCAGGACCTTGCGGGG-3′。
荧光引物和荧光底物均从深圳华大基因股份有限公司订购合成。
1.1.3 仪器
AKTA avant 25蛋白纯化系统(GE Healthcare Life Sciences),Typhoon扫描仪(GE Healthcare Life Sciences),垂直电泳仪Mini-PROTEANR Tetra Handcast System [伯乐生命医学产品(上海)有限公司],JS-3000凝胶成像仪(上海培清科技有限公司),Nano Drop微量核酸蛋白测定仪(ThermoFisher Scientific),恒温摇床(上海知楚仪器有限公司),Biosafer超声破碎仪[赛飞(中国)有限公司],高压均质机[永联生物科技(上海)有限公司],100 L自控式发酵罐(镇江格瑞生物工程有限公司),Avanti J-26冷冻离心机(Beckman coulter)。
1.2 方法
1.2.1 重组蛋白表达
表达质粒用热激法转化EscherichiacoliBL21(DE3)感受态细胞,转化液涂布到含100 μg/mL氨苄青霉素(Ampicillin,Amp)的LB平板,37℃倒置培养过夜。
Pol D DNA聚合酶小亚基DP1重组菌的表达:挑取单菌落在LB/Amp液体培养基中培养两级种子,以2%的接种量转接到3瓶2.5 L的LB/Amp液体培养基,37℃、150 r/min摇床培养6-8 h,待其OD600值达1.0时,加入终浓度为50 mmol/L的IPTG诱导培养8-10 h,离心收集菌体,-80℃冻存。
Pol D DNA聚合酶大亚基DP2重组菌的表达:由于DP2表达量较低,因而采用发酵罐进行大量培养;挑取单菌落在 LB/Amp液体培养基中培养三级种子,以1%的接种量转接到100 L LB/Amp液体培养基的发酵罐中,在温度37℃、转速120 r/min、通气量为0.5 vvm、罐压0.05 MPa的条件下发酵培养,待其OD600值达到1.0时,加入终浓度为50 mmol/L的IPTG继续培养8-10 h诱导蛋白表达,用管式离心机离心收集菌体,-80℃冻存。
1.2.2 重组蛋白纯化
(1)Ni-NTA柱层析。
取10 g表达Pol D DNA聚合酶小亚基DP1的菌泥重悬于50 mL Ni-NTA Binding Buffer,置70℃水浴热处理20 min,使大部分非耐热蛋白失活沉淀;用超声波破碎仪破碎菌体后,于4℃、13 000 r/min离心40 min,收集上清;取160 g表达Pol D DNA聚合酶大亚基DP2的菌泥重悬于500 mL Ni-NTA Binding Buffer,置70℃水浴热处理20 min后,用高压均质机在4℃下高压研磨破碎菌体约10 min,4℃、13 000 r/min离心40 min,收集上清。
在AKTA avant25蛋白纯化系统上进行Ni-NTA柱层析纯化,将细胞破碎液上清与5 mL平衡好的Ni-NTA树脂结合,采用3倍柱体积的Binding Buffer清洗后,用咪唑浓度为0-250 mmol/L的线性梯度液进行洗脱,洗脱液按20 mL/管进行收集并置4℃冰箱暂存;取少量洗脱液进行SDS聚丙烯酰胺凝胶电泳,并用ImageJ软件分析计算每管洗脱液中目的蛋白条带的占比,根据电泳结果选取含有目的蛋白的样品,用强阴离子吸附柱进一步纯化。
(2)强阴离子吸附柱(Q柱)层析。
将经Ni-NTA纯化后的含有目的蛋白的组分混合,用含25 mmol/L Tris (pH值为8.0)溶液进行降盐透析24 h,每6 h换一次透析液,使样品中NaCl浓度降至10 mmol/L以下;取透析完成的样品与5 mL Q阴离子交换树脂结合,用NaCl线性梯度洗脱液进行升浓度梯度洗脱,NaCl线性梯度液浓度为0-1 000 mmol/L,洗脱液按10 mL/管收集并置4℃暂存;取少量洗脱液样品进行SDS聚丙烯酰胺凝胶电泳,根据电泳结果选取含有目的蛋白的样品,分别用DEAE柱(DEAE树脂)或疏水柱(Butyl树脂)进一步纯化。
(3)弱阴离子交换柱层析
将经Q柱纯化后的含有重组Pol D DNA 聚合酶小亚基DP1的样品混合,按(2)的方法透析;取透析完成的样品与5 mL DEAE阴离子交换树脂结合,用NaCl线性梯度洗脱液进行升浓度梯度洗脱,NaCl梯度液浓度为0-1 000 mmol/L,洗脱液按5 mL/管收集并置4℃冰箱暂存;取少量洗脱液样品进行SDS聚丙烯酰胺凝胶电泳。
(4)疏水柱层析。
将Q柱纯化后含有重组Pol D DNA聚合酶大亚基DP2的样品组分混合,计算并加入5 mol/L NaCl溶液,使样品中NaCl浓度达到3.5 mol/L;将此样品溶液与用3.5 mol/L NaCl溶液平衡过的5 mL Butyl树脂结合,用NaCl线性梯度洗脱液进行降浓度梯度洗脱,NaCl梯度液浓度为3 500-0 mmol/L,洗脱液按5 mL/管收集并置4℃暂存;取少量洗脱液样品进行SDS聚丙烯酰胺凝胶电泳。
(5)蛋白的透析与保存。
将纯化后的目的蛋白样品混合,用含25 mmol/L Tris (pH值为8.0)、0.1 mmol/L EDTA、200mmol/L KCl、1 mmol/L DTT和50%甘油的Storage Buffer在4℃透析18 h,每6 h换一次Storage Buffe,透析完毕用微量核酸蛋白测定仪测定蛋白质浓度,置-20℃保存。
1.2.3 重组Pol D DNA聚合酶大小亚基的功能实验
(1)重组Pol D DNA聚合酶大亚基DP2的延伸活性。
根据单链DNA模板ZXS001的序列设计和合成带有5′-Cy3荧光标记的引物Cy3-ZXS002,配制荧光引物-模板混合物(100 pmol Cy3-ZXS002、200 pmol ZXS001、7 mmol/L pH 值为8.0的Tris、50 mmol/L NaCl、7 mmol/L MgCl2),沸水浴5 min后自然冷却至室温,使荧光引物互补配对结合到单链DNA模板;配制10 μL延伸反应体系:20 mmol/L pH 值为8.0的Tris、10 mmol/L MgSO4、10 mmol/L KCl、10 mmol/L (NH4)2SO4、0.1% Trition X-100、0.5 mmol/L dNTP、0.8 μL荧光引物-模板混合物、1 μL 不同浓度(0 pmol、0.1 pmol、0.2 pmol、0.5 pmol、1.5 pmol、5 pmol、15 pmol、40 pmol)的DP2或DP2与DP1的混合物,在70℃孵育2 min使荧光引物发生DNA复制延伸反应;加入等体积的2×Stop Buffer (90%甲酰胺、10 mmol/L EDTA、10% TBE)终止反应,接着煮沸5 min,冰水浴使其快速冷却,此时未经过延伸的荧光引物会与单链DNA模板分离,游离于体系内;延伸反应物在含8 mol/L尿素的TBE溶液中进行聚丙烯酰胺凝胶电泳,最后用GE Typhoon扫描仪Cy3频段扫描检测。
(2)重组Pol D DNA聚合酶小亚基DP1外切酶活性。
合成5′-Cy5荧光标记的单链DNA底物Cy5-0A01,构建20 μL降解反应体系:50 mmol/L pH 值为8.0的Tris,4 mmol/L MgCl2、100 μg BSA、2 pmol Cy5-0A01、1 μL 不同浓度(0 pmol、1.562 5 pmol、3.125 pmol、6.25 pmol、12.5 pmol、25 pmol、50 pmol、100 pmol)的DP1或DP1与DP2的混合物或DP1与GINS 51的混合物,ddH2O补足到20 μL。反应体系在70℃孵育一定时间(0 s、30 s、1 min、2 min、5 min、10 min、15 min、20 min)后加入等体积的2×Stop Buffer中止反应,煮沸5 min,待其冷却后进行聚丙烯酰胺凝胶电泳,最后用GE Typhoon扫描仪Cy5频段进行扫描检测。
2 结果与分析
2.1 重组Pol D DNA聚合酶大小亚基的纯化
重组Pol D DNA聚合酶小亚基DP1先后经过Ni-NTA柱、Q柱和DEAE柱层析纯化,DP1在每次层析的收集组分中的占比依次增加。收集合并DEAE柱层析中DP1占比分别为77.9%、95.3%和98.6%的第5-7管组分,经Storage Buffer透析后收得含DP1蛋白溶液22.5 mL,微量核酸蛋白测定仪测定其蛋白质浓度为1.49 mg/mL。
重组Pol D DNA聚合酶大亚基DP2先后经过Ni-NTA、Q柱和Butyl疏水柱层析纯化,DP2在每次层析的收集组分中的占比依次增加。收集合并Butyl疏水层析后DP2占比分别为70.5%、82.4%、81.2%、91.5%、90.6%、79.0%的第11-16管组分,经Storage Buffer透析后收得含DP2蛋白溶液11 mL,微量核酸蛋白测定仪测定其蛋白质浓度为0.56 mg/mL。
将以上较高纯度的DP1蛋白和DP2蛋白用于研究其功能。
2.2 重组Pol D DNA聚合酶大小亚基的功能
2.2.1 重组Pol D DNA聚合酶大亚基DP2的延伸活性
在延伸反应体系中分别加入0-40 pmol的DP2蛋白反应2 min后,其聚丙烯酰胺凝胶电泳结果如图1所示。由图1可知,含有DP2蛋白的实验样品完成了荧光核酸引物的复制延伸,DP2蛋白为零的对照样品未完成荧光核酸引物的复制延伸,说明重组Pol D DNA聚合酶大亚基DP2具有核酸复制延伸活性,且延伸活性较强,在用量低至0.1 pmol时仍可完成荧光核酸引物的复制延伸。
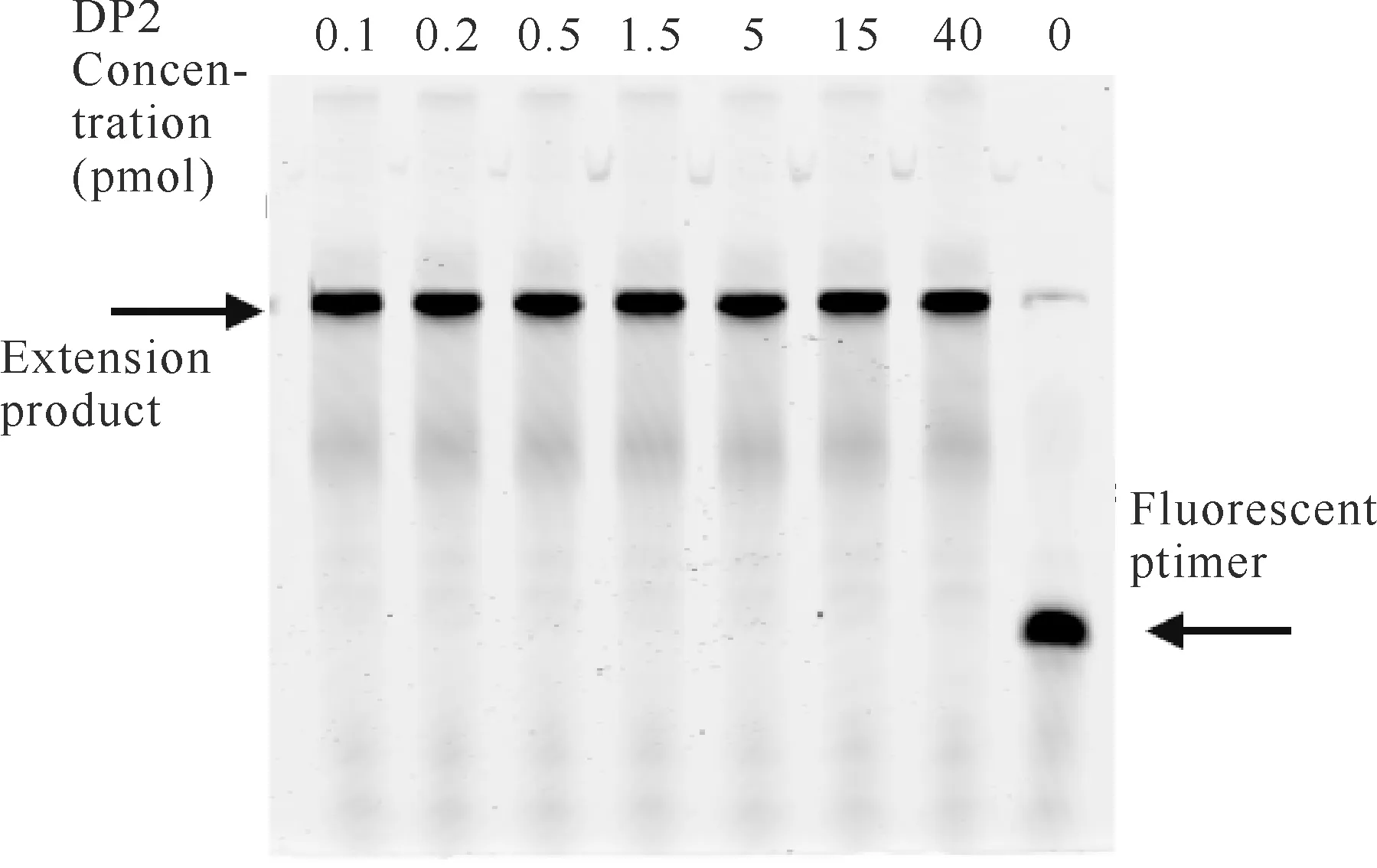
图1 DP2浓度对其延伸活性的影响
2.2.2 重组Pol D DNA聚合酶小亚基DP1的外切酶活性
在降解反应体系中分别加入0-100 pmol的DP1蛋白降解反应15 min后,其聚丙烯酰胺凝胶电泳结果如图2所示。由图2可知,加入DP1蛋白后荧光核酸底物被降解而逐渐变短,且在相同反应时间内,随着DP1用量的增加,荧光核酸底物被降解得更短,表明Pol D DNA聚合酶小亚基DP1具有3′→5′外切核酸酶活性。

图2 DP1浓度对其3′→5′外切核酸酶活性的影响
在降解反应体系中DP1蛋白用量为50 pmol 的条件下,经不同降解反应时间后,其聚丙烯酰胺凝胶电泳结果如图3所示。由图3可知,随着反应时间的增加,DP1对荧光核酸底物的降解程度逐渐增大。

图3 DP1反应时间对DP1降解核酸底物的影响
2.2.3 重组Pol D DNA聚合酶小亚基DP1对大亚基DP2延伸活性的影响
在延伸反应体系中DP2蛋白用量为5 pmol 的条件下,分别加入0-40 pmol的DP1蛋白,延伸反应2 min后,其聚丙烯酰胺凝胶电泳结果如图4所示。由图4可知,在DP1含量较低时,DP2的延伸活性仍可以使DNA链正常复制延伸,但DP1用量达到1.5 pmol时(第4泳道)已开始导致延伸链被降解,出现游离的荧光分子;当DP1与DP2的量相当时(第5泳道),DP2延伸的DNA开始明显被降解,但仍有完整的延伸链保留;当DP1用量大于DP2时(第6,7泳道),体系内延伸复制的DNA和荧光引物都被降解。因此可以推测,至少在细胞外体系中,在Pol D DNA聚合酶执行复制功能时,复制效率达到最高时的小亚基DP1与大亚基DP2的最佳浓度比低于0.3∶1,并且至少在DP1的量提高到与DP2的量相当时(即1∶1),就有必要使用其他的功能蛋白来抑制小亚基DP1的外切酶活性。

图4 DP1浓度对DP2延伸活性的影响
2.2.4 重组Pol D DNA聚合酶大亚基DP2对小亚基DP1外切酶活性的影响
在降解反应体系中DP1蛋白用量为20 pmol 的条件下,分别加入0-100 pmol 的 DP2蛋白,降解反应2 min后,其聚丙烯酰胺凝胶电泳结果如图5所示。由图5可知,在无DP1的情况下,DP2自身不存在外切核酸酶活性,但DP2对DP1的外切核酸酶活性有影响,在相同反应时间内,随着DP2的增加,DP1对荧光核酸底物的降解更彻底,说明DP2对DP1的外切核酸酶活性具有促进作用。

图5 DP2浓度对DP1降解核酸底物的影响
在降解反应体系中DP1蛋白用量为50 pmol的条件下,添加50 pmol的DP2蛋白,经不同降解反应时间后,其聚丙烯酰胺凝胶电泳结果如图6所示。图6与图3比较可知,在DP2存在时DP1降解荧光核酸底物所需的时间明显缩短,10-15 min已基本降解完毕,也进一步表明DP2具有促进DP1外切核酸酶活性的作用。
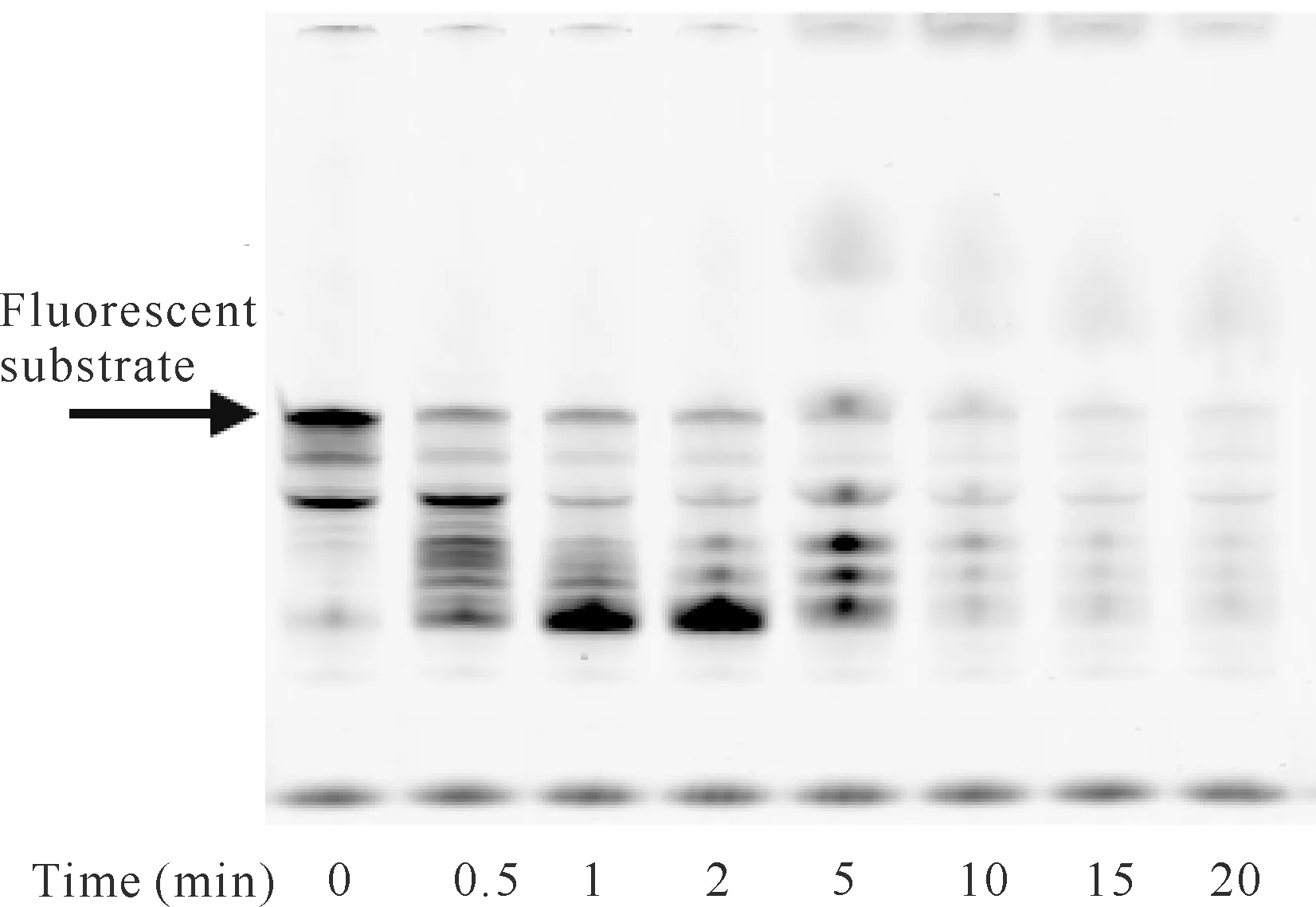
图6 DP2存在时反应时间对DP1降解核酸底物的影响
2.2.5 GINS 51蛋白对重组Pol D DNA聚合酶小亚基DP1外切酶活性的影响
古菌的GINS四聚体会与Cdc45、MCM(Minichromosome Maintenance)、Pol D、PCNA等蛋白发生相互作用[12-15],是古菌细胞存活必需的蛋白复合物。在降解反应体系中DP1蛋白用量为5 pmol的条件下,分别加入0-80 pmol的Thermococcussp.4557 GINS 51蛋白,降解反应2 min后,其聚丙烯酰胺凝胶电泳结果如图7所示。由图7可知,在无DP1的情况下,GINS 51自身不存在外切核酸酶活性,但GINS 51对DP1的外切核酸酶活性有影响,在相同反应时间内,随着GINS 51的增加,DP1对荧光核酸底物的降解呈减弱趋势,说明GINS 51对DP1的外切核酸酶活性有抑制作用。
在降解反应体系中DP1蛋白用量为50 pmol的条件下,添加50 pmol的GINS 51蛋白,经不同降解反应时间后,其聚丙烯酰胺凝胶电泳结果如图8所示。图8与图3比较可知,相同反应时间内,加入GINS 51后DP1降解核酸底物的条带略有减少,也说明GINS 51对DP1的外切核酸酶活性有一定抑制作用。
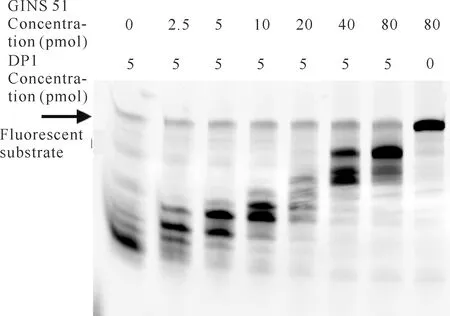
图7 GINS 51浓度对DP1降解核酸底物的影响

图8 GINS 51存在时反应时间对DP1降解核酸底物的影响
3 讨论
本研究发现,纯化的重组Thermococcussp.4557 Pol D DNA聚合酶大亚基DP2蛋白具有DNA复制延伸活性,小亚基DP1蛋白具有3′→5′外切核酸酶活性,这与此前人们对Pol D DNA聚合酶的认识一致[16]。在DNA复制过程中,DNA聚合酶会对复制错误的片段进行降解,以保证DNA复制的准确性。有研究表明Pol D DNA聚合酶是由小亚基DP1和大亚基DP2以1∶1形成的二聚体[17],本研究发现Thermococcussp.4557 Pol D DNA聚合酶小亚基DP1的3′→5′外切核酸酶活性较强,在与大亚基DP2以1∶1的比例结合时,大亚基DP2复制延伸的DNA被明显降解,表明单独按1∶1形成的小亚基DP1和大亚基DP2二聚体无法完成DNA的复制延伸;由于GINS 51对小亚基DP1的外切核酸酶活性有抑制作用,因此推测Pol D DNA聚合酶完成DNA的复制延伸功能时可能有GINS 51参与调控小亚基DP1的外切核酸酶活性。Lu等[18]对Thermococcussp.4557 Pol D DNA聚合酶与GINS、GAN等蛋白进行了分子筛互作分析,并提出了名为Pol D-GINS-GAN的古菌DNA复制复合体模型;该研究表明,单独的Pol D DNA聚合酶大亚基DP2与GINS、GAN等蛋白不发生互作,而单独的Pol D DNA聚合酶小亚基DP1可以与GINS 51发生互作,并且单独的GINS 51与GAN发生互作,因此大亚基DP2需要借助小亚基DP1才能与复制叉上的复制复合体连接。结合本研究结果,可以进一步推测Pol D DNA聚合酶在DNA复制过程中的功能与机理:GINS 51与小亚基DP1结合在一起,通过DP1再与大亚基DP2连接;大亚基DP2与DNA底物结合并行使复制延伸功能;GINS 51通过抑制小亚基DP1的3′→5′外切核酸酶活性来保证大亚基DP2可以顺利进行新链的复制延伸;同时,当大亚基DP2正在合成的新链出现错配时,小亚基DP1的3′→5′外切核酸酶活性被激活,迅速切除错配片段,从而保证DNA复制顺利进行。DNA复制错配发生时GINS 51对小亚基DP1的3′→5′外切核酸酶活性的抑制作用如何解除,复制过程中GAN、PCNA、SSB等其他复制因子、调控因子如何结合并参与调控,仍然有待进一步研究。
4 结论
利用荧光标记核酸引物进行的DNA复制延伸实验和荧光标记核酸底物进行的DNA降解实验表明,纯化的重组Thermococcussp.4557 Pol D DNA聚合酶大亚基DP2蛋白具有DNA复制延伸活性;纯化的重组Thermococcussp.4557 Pol D DNA聚合酶小亚基DP1蛋白具有3′→5′外切核酸酶活性。DP2蛋白的延伸活性受到DP1蛋白的影响,若DP1与DP2浓度比例小于0.3∶1时,DP2能完成荧光核酸引物的延伸;DP1提高到与DP2含量相当(1∶1)时,出现明显的延伸产物和荧光引物降解现象;DP1与DP2的浓度比进一步提高到3∶1时,可将延伸产物和荧光引物完全降解。Thermococcussp.4557的DP2蛋白和复制相关蛋白GINS 51对DP1蛋白的外切核酸酶活性有不同的作用,DP2蛋白对DP1蛋白的外切核酸酶活性有促进作用,而GINS 51对DP1蛋白的外切核酸酶活性有抑制作用。
