JAK1抑制剂Upadacitinib(Rinvoq)用于治疗类风湿性关节炎的研究进展
2022-02-08王小涵代洪爽何梓煊张荠方
陈 烨,王 铭,王小涵,代洪爽,丁 实,王 洋,何梓煊,张荠方,刘 举
(辽宁大学 药学院,辽宁 沈阳 110036)
0 引言
类风湿性关节炎(Rheumatoid arthritis,RA)是一种以慢性炎症滑膜炎和进行性关节破坏为特征的系统性自身免疫性疾病[1].全世界患病率为0.5%~1%[2].虽然RA的病因尚不清楚,但一些遗传多态性和环境因素与增加的易感性和疾病严重程度相关[3].RA主要影响周围关节、滑膜组织的异常炎症增生导致软骨损伤和骨侵蚀[4-5].此外,RA的慢性全身炎症也可导致关节外表现的发展,如慢性贫血、疲劳和肺间质疾病,并可导致共病,如骨质疏松、感染、癌症、增加心血管疾病、Ⅱ型糖尿病和心理障碍[6-7].因此,RA的特征是随时间推移逐渐丧失能力,并且与普通人群相比,增加死亡风险[8].这些病理过程涉及炎症细胞因子和自身反应性T细胞.在过去的几十年中,通过引入一种治疗目标的方法来防止关节进一步损伤,RA的管理得到了显著的改善,但仍有相当比例的RA患者未能达到临床目标,因此,最近的研究重点已转向抑制参与炎症信号传导到免疫细胞的激酶.Janus激醇(Janus kinase,JAK)在引发RA和其他自身免疫性疾病的症状中起着关键作用,因为它们的磷酸化通常是细胞因子激活的细胞信号通路的一部分.JAK激酶有4种(JAK1、JAK2、JAK3和TYK2),每种JAK在控制各种免疫反应中都具有高度特异性的功能.特别是Baricitinib和Tofacitinib两种JAK抑制剂,在不同RA亚群中进行随机对照试验(RCT),发现它们对JAK家族的大多数成员(JAK1、JAK2、JAK3和TYK2)都有活性,目前已经被批准用于RA的治疗.最近研究者主要开发针对JAK1选择性抑制剂(Upadacitinib),目的是通过降低对JAK3和JAK2的影响来提高其安全性.该分子是一种选择性JAK1抑制剂(JAK1的IC50:0.043 μmol/L;JAK2:0.2 μmol/L;JAK3:2.3 μmol/L;TYK2:4.7 μmol/L),在各种自身免疫性疾病患者的临床试验中显示出较好的疗效、安全性和耐受性[9-10].
Upadacitinib(Rinvoq)是一种口服JAK1抑制剂,由AbbVie开发用于治疗RA和其他免疫性炎症疾病.JAK激酶(JAK1、JAK2、JAK3和TYK2)在多种细胞内细胞因子信号传导中发挥作用,JAK介导的信号传导途径在正常状态和免疫性炎症疾病(包括类风湿性关节炎)的病理状态中都很重要[11].2019年8月16日,Upadacitinib作为高选择性JAK1抑制剂,在美国获得批准用于中等至严重的RA以及有或不耐受反应不足的甲氨蝶呤的Ⅲ期临床试验项目[12].Upadacitinib为缓释片(ER),每片15 mg,推荐剂量为15 mg,每日口服一次;它可以作为单一疗法或与甲氨蝶呤等其他非生物疾病修饰抗风湿药物(DMARDs)联合使用[13].由于RA发病机制尚未完全明确,本文将从Upadacitinib合成、有效性、药代动力学和毒副作用等4个方面进行综述,以期为RA治疗寻求新的突破.
1 Upadacitinib的合成
Abbott Laboratories的专利首先阐述了Upadacitinib的结构及其合成的信息,但是由于子步骤中并没有详细描述,因此合成路径重建困难[14].随后的一项AbbVie专利虽然对用户更有一定参考价值,但其中的一些模糊的描述也缺乏应用性(例如,最好环境温度为60 ℃左右)[15].然而,2017年AbbVie专利则对合成途径给出了较为详细的描述[16].首先,化合物1用于氨基甲酸乙酯的芳基化合成化合物2(见图1).然后由Cbz保护的甘氨酸酯3合成了分子中含有吡咯烷酮的部分.化合物3与丙烯酸乙酯(共轭加成,然后分子内进行克莱森缩合)反应生成高度烯醇化的酯中间体4.经过磺酰化后,Suzuki交叉偶联引入一个乙基,生成6.酯水解后,对映体选择性加氢和添加二环己胺得到盐8,见图2.
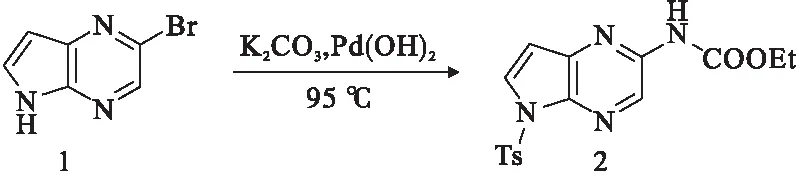
图1 氨基甲酸酯2的制备[16]
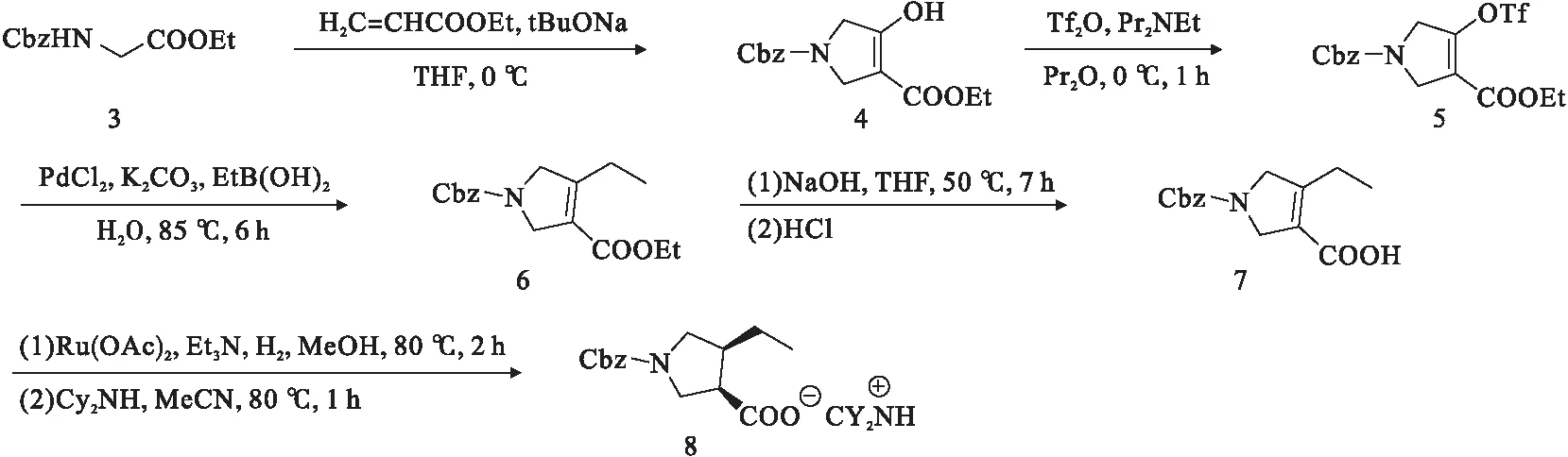
图2 二环己胺盐8的制备路线图[16]
图3中用(R)-1-(萘酰基)乙胺盐8(8和9具有相同的羧酸盐阴离子)继续合成,从9得到吡咯烷羧酸10,在亚砜的作用下将羧基—COOH基团转化为—COCH2Br基团,所得化合物12用于氨基甲酸酯2的氨基甲酸酯部分的N-烷基化[17],然后经过三氟乙酸酐环化和Cbz脱保护(氢解)[18],最后利用CDI和2,2,2-三氟乙胺[19]来合成目标化合物[20].Srinivas等[21]在2019年申请的一项专利中发现了其他合成途径.
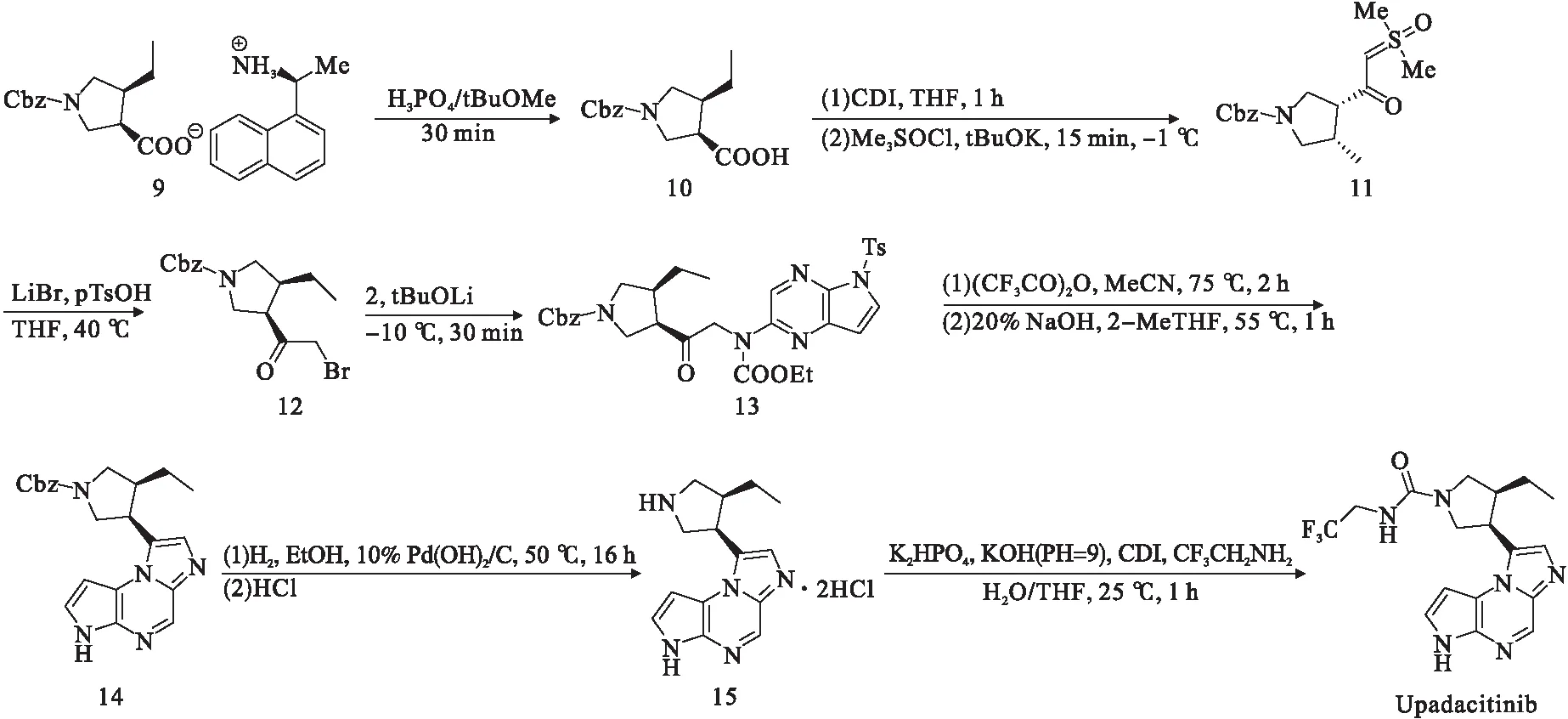
图3 化合物9合成Upadacitinib(Rinvoq)路线图[17-20]
2 Upadacitinib的有效性研究
Upadacitinib对JAK1的选择性高于JAK2、JAK3和TYK2(在生化分析中IC50分别为0.045、0.109、2.1、4.7 μmol/L,在细胞分析中IC50分别为0.014、0.593、1.860、2.715 μmol/L)[22].因此,Upadacitinib有可能选择性地抑制与JAK1依赖性途径相关的胞质分裂信号(如与RA相关的IL-6和IFNγ信号),同时能减少涉及JAK2(促红细胞生成素(EPO)受体(造血)信号)和JAK3(例如IL-15(NK细胞稳态)信号)的生理功能对细胞因子信号的影响.Parmentier等[22]研究表明,Upadacitinib能有效抑制JAK1依赖性细胞因子IL-6、肿瘤抑制素M、IL-2和IFNγ,其活性是促红细胞生成素信号的60倍.此外,Upadacitinib对人全血IL-6信号的抑制作用也很明显(CD3+T细胞和CD14+单核细胞群IC50分别为0.207和0.078 μmol/L).
健康志愿者服用Upadacitinib缓释片后,导致全血中STAT3和STAT5的磷酸化(分别由IL-6和IL-7诱导)会受到Upadacitinib剂量和浓度的依赖性抑制,其最大抑制发生在治疗1 h后,并在治疗间隔结束时恢复到接近基线的水平[13].研究者通过对参与第Ⅲ阶段SELECT-NEXT(NCT02675246)和SELECT-BEYOND(NCT02706847)试验的RA患者血浆蛋白进行分析[23],发现跟RA病理生物学相关的Upadacitinib标准化途径(IL-1、IL-6、IL-12、IL-15、IL-18、IFNα、IFNβ、IFNγ、CSF2和TNF)和白细胞活性(包括白细胞迁移、T细胞反应和炎症反应).
2.1 大鼠模型
在大鼠佐剂性关节炎和胶原性关节炎模型中(两者都有显著的IL-6和IFNγ表达),Upadacitinib 3 mg/kg和10 mg/kg可改善滑膜肥大、发育、软骨损伤和骨侵蚀.虽然Upadacitinib和Tofacitinib(一种Pan-JAK抑制剂)在关节炎大鼠模型中显示出有效性,但Upadacitinib在网织红细胞分布和循环自然杀伤(NK)细胞数量方面的疗效不如Tofacitinib[22].
2.2 临床试验
在Upadacitinib治疗类风湿性关节炎的临床试验中[24],研究者观察到血脂(低密度脂蛋白胆固醇(LDL-C)和高密度脂蛋白胆固醇(HDL-C))水平与 C反应蛋白水平成反比关系,尤其对治疗有持续反应的患者相关性最强.Sokolove等[25]研究表明,Upadacitinib治疗显著增加RA患者依赖性胆固醇(ABCA-1)排出能力,并且胆固醇逆向转运的改善与C反应蛋白(CRP)水平的降低以及总胆固醇和HDL-C水平的变化相关.
表1为Upadacitinib在Ⅲ期临床试验的评估概况.从表中可以看出,该评估可分为5个关键性试验:1)未使用甲氨蝶呤治疗的RA患者;2)甲氨蝶呤治疗反应不充分(SELECT-MONOTHERAPY,n=433)[26]的中度至重度活动期RA患者,大部分患者未使用其他cDMARDs(SELECT-EARLY,n=631)[27];3)cDMARD治疗不理想后与安慰剂的附加治疗(SELECT-NEXT,n=442)[28];4)阿达木单抗(Adalimu-mab)联合甲氨蝶呤治疗(SELECTCOMPARE,n=1 629)[29];5)bDMARD治疗失败后与安慰剂联合cDMARD治疗(SELECT-BEYOND,n=333)[11].以上数据不包括随机接受服用30 mg Upadacitinib治疗的患者(其疗效稍高,但未获得批准[30]).

表1 每天15 mg Upadacitinib第3阶段试验的主要终点
从表1还可以看出,主要终点为第12周[11,28]和第14周[27],疾病活动度低并在第12周[29]和第24周[26]缓解.在未接受甲氨蝶呤治疗的患者中,近半数患者在6个月后缓解,而接受甲氨蝶呤治疗的患者中约为五分之一在6个月后也达到缓解状态[26].与持续服用甲氨蝶呤的患者相比,在14周后改用Upadacitinib的患者在疾病活动度较低时的反应率有类似的差异;同样,在第5周和第12周时Upadacitinib与cDMARD联合使用的患者与单独使用安慰剂的患者相比,在疾病活动度较低时的反应率也有类似的差异[11,28].对于单独使用甲氨蝶呤不足的患者,在12周开始服用Upadacitinib、Adalimu-mab或安慰剂后,其缓解率分别为29%、18%和6%[29].在48周的全部治疗过程中,与使用Adalimu-mab的患者相比,使用Upadacitinib的患者表现出较低的疾病活动性和缓解率[31],Upadacitinib具有更好的临床表现.
上述差异与次要终点(包括美国风湿病学会(ACR)20、50和70反应以及临床疾病活动指数)的持续改善有关.在使用Upadacitinib和Adalimu-mab 48周后,对比使用安慰剂组发现其放射性疾病进展患者的比例均显著降低[31].以上情况表明Upadacitinib在SELECT-BEYOND方面(bDMARD失败后以及与cDMARD联合用药后)显著地改善了患者的疼痛和僵硬状况[32].
3 Upadacitinib的药代动力学研究
口服Upadacitinib的药代动力学曲线的剂量范围为IR制剂1~48 mg[33],ER制剂7.5~30 mg[34].在Upadacitinib的群体药代动力学模型中,估算ER制剂相对于IR制剂的口服生物利用度为76.2%[34].对于RA患者在重复服用Upadacitinib后,患者体内IR制剂的血浆浓度达到峰值的中位时间为12 h[10],ER制剂的血浆浓度达到峰值的中位时间为24 h[35].在禁食条件下,每天多次服用15和30 mg ER Upadacitinib后的24 h内稳态血药浓度时间曲线下面积值(AUC24)与每天多次服用6和12mg IR Upadacitinib后的AUC24值相等[35].每日一次重复给药Upadacitinib后在4 d内达到的稳态血浆浓度[36];每日一次重复给药后的累积量最小[13,35].Upadacitinib是中等(52%)的血浆蛋白结合物,在血浆和血细胞成分(血液和血浆容积之比为1∶0)之间的分配相似.服用高脂肪/高热量膳食的Upadacitinib对Upadacitinib暴露没有临床相关影响[35-36],并且该药物可与食物一起服用也可以不与食物一起服用[13].
3.1 代谢
Upadacitinib主要由细胞色素P450(CYP)同工酶CYP3A4代谢,其主要代谢物是葡萄糖醛酸化后的单氧化产物(单剂量14C Upadacitinib IR溶液占血浆放射性总量的13%).由于未检测到活性代谢物,因此Upadacitinib 由CYP2D6代谢的可能性很小[13].Upadacitinib的药理活性是由母体分子引起的,占血浆总放射性的79%.未代谢的Upadacitinib通过尿液和粪便直接排出体外,两种途径所占比例分别为24%和38%,而34%Upadacitinib作为代谢物消除.Upadacitinib的平均终端消除半衰期为8~14 h[13].
体重、性别、种族、年龄、肾功能损害(轻度、中度或重度[37])和轻度或中度(Child-Pugh A或B[38])肝功能损害对Upadacitinib暴露无临床相关影响[13].Upadacitinib尚未在严重(Child-Pugh-C)肝损害患者中进行研究[13].
3.2 药物合用
当与强CYP3A4抑制剂(如酮康唑)合用时,Upadacitinib血浆暴露量增加;当与强CYP3A4诱导剂(如利福平)合用时,Upadacitinib血浆暴露量减少[35].在接受强CYP3A4抑制剂慢性治疗的患者中,应谨慎使用Upadacitinib.联合使用Upadacitinib和强CYP3A4诱导剂可能会降低Upadacitinib的疗效,因此不推荐使用[13].预计CYP2D6抑制剂不会对Upadacitinib血浆暴露有临床相关影响[13,33].甲氨蝶呤与Upadacitinib合用对Upadacitinib血浆暴露也无影响[10].
群体药代动力学分析和体外研究表明,改变pH值的药物(如抗酸剂或质子泵抑制剂)预计不会影响Upadacitinib的暴露[13,34].体外评估表明,在临床相关浓度下,Upadacitinib不是CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2D6和CYP3A4的诱导物或抑制剂,并且不抑制转运蛋白P-gp、BCRP、OATP1B1、OATP1B3、OCT1、OCT2、OAT1、OAT3、MATE1和MATE2 K[13].在临床研究中,Upadacitinib对合用药物的药代动力学无临床相关影响[13,39].
4 毒副作用
Upadacitinib常见的不良反应是上呼吸道感染、恶心、尿路感染、鼻咽炎、丙氨酸氨基转移酶(ALT)升高、磷酸肌酸激酶(CPK)升高和咳嗽,此外中性粒细胞减少和高胆固醇血症(总胆固醇和低密度脂蛋白胆固醇平均增加13%,甘油三酯平均增加10%)很常见[30].严重感染的频率为每100个患者每年3.6次,与其他JAK抑制剂的感染频率相似[30].与Upadacitinib相关的机会性感染的发生率为每100名患者每年发生0.6次[30].
表2为Upadacitinib治疗期间TEAE的暴露调整事件发生率.从表2中可看出,Upadacitinib的带状疱疹(HZ)事件发生率高于Adalimu-mab.无HZ事件发生主要是脑膜脑病或非皮肤内脏器官.除了Upadacitinib的一个事件报告为眼科并导致研究药物停药,大多数HZ事件并不严重,仅涉及1~2个皮节.然而,Upadacitinib恶性肿瘤的事件发生率和Adalimu-mab相似,未观察到明显的恶性肿瘤模式或类型.NMSCs是Upadacitinib治疗中出现的两种基底细胞癌和一种鳞状细胞癌(没有报告治疗中出现的淋巴瘤病例).Upadacitinib的第3个事件被归类为“胃肠(GI)穿孔”,这些事件并不是自发性GI穿孔,而是在输卵管脓肿、肛门脓肿和肛瘘的情况下发生腹膜炎和阑尾炎事件.虽然大多数有关肝脏事件的报告为无症状且ALT和谷草转氨酶(AST)升高,但不会导致过早停用研究药物.
在Upadacitinib治疗组的4个死亡病例中,一名患者患有深静脉血栓形成(DVT),两名患者患有肺栓塞(PE),一名患者患有DVT和PE,以上4个死亡病例患者都具有心血管病所引发死亡的已知危险因素.总体而言,持续接受Upadacitinib治疗的患者在第48周的血红蛋白、中性粒细胞、淋巴细胞和血小板的平均水平,与前26周保持一致且在正常范围内.尽管LDL-C、HDL-C、总胆固醇和HDL-C的比率保持稳定,但在Upadacitinib治疗组中均观察到LDL-C和HDL-C升高的情况.另外,Upadacitinib和Adalimu-mab两个治疗组中均出现少数患者的实验室参数变化大于或等于3级,这些参数包括血红蛋白、中性粒细胞和血小板计数降低.与Adalimu-mab治疗组相比,Upadacitinib治疗组中出现3级和4级淋巴细胞减少的患者比例更高,但未发现低淋巴细胞计数、低中性粒细胞计数与感染率(包括严重和机会性感染)、HZ之间的关联性.ALT/AST 3/4级升高的情况并不常见,但在Upadacitinib治疗组中的发生率高于Adalimu-mab或安慰剂的治疗组.在Upadacitinib治疗组中会有大于或等于3级CPK升高的病例,例如Upadacitinib治疗组中的两名患者(一名肌肉无力,一名肌肉疼痛)在剧烈活动后出现单次3级CPK升高的情况,除上述两个病例外,Upadacitinib治疗组中大多数患者无症状.在没有中断Upadacitinib药物的情况下,还有两名患者的CPK虽然升高但随后患者病情又趋向正常化.此外,Upadacitinib治疗组中没有出现横纹肌溶解或因CPK升高而停药的病例.总之,Upadacitinib治疗组中因不良反应而停药的病例发生的平均比率为2.8%,而安慰剂治疗组中的平均比率为2.0%[30].
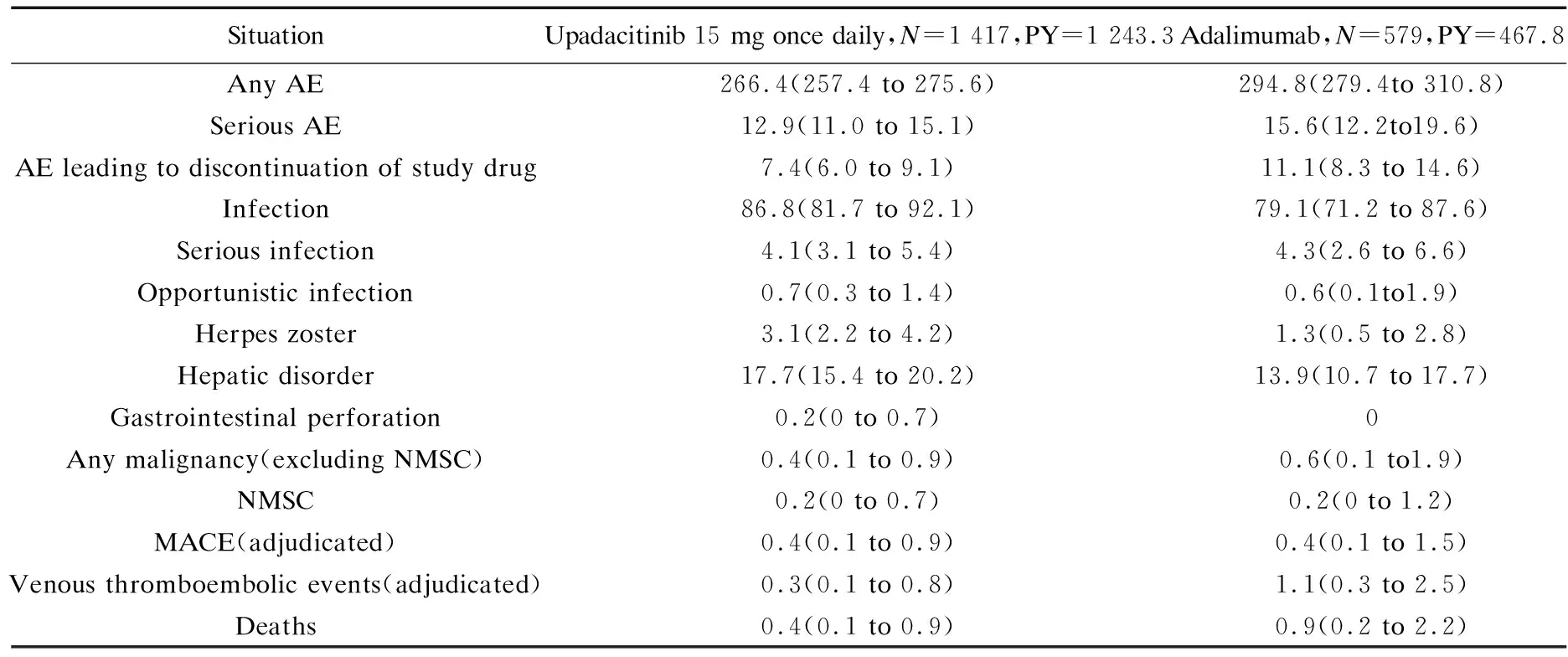
表2 TEAE 的暴露调整事件发生率(E/100 PYs(95% Cl))
5 展望
RA是一种慢性自身免疫性疾病,其发病机制尚未完全明确,对其治疗也一直困扰着很多医药工作者.近年来,大量的研究表明JAK抑制剂对于治疗RA有着很明显的效果,为治疗RA提供了一个重要的研究方向.虽然Upadacitinib作为JAK1抑制剂对RA有着良好的效果,但也有感染的副作用.此外,JAK抑制剂还可以治疗银屑病、溃疡性疾病及非小细胞肺癌等[40],所以Upadacitinib还有广泛的研究价值和应用前景.
