PCR-核酸试纸条快速鉴定皮氏罗尔斯顿菌
2022-02-03赵潇颖姜菲菲孙丽颖宿建胜赵云冬
王 丹,赵潇颖,姜菲菲,孙丽颖,许 萌,宿建胜,赵云冬
北华大学医学技术学院,吉林 吉林 132013
制药用水是药品生产中最重要的原料,制药用水的质量直接影响药品的质量,尤其是灭菌的注射用水。灭菌的注射用水常用于灭菌粉末的溶剂或注射剂的稀释剂,直接注射到人体组织中,关乎患者的生命安全[1]。2015年《中华人民共和国药典》明确规定灭菌注射用水为注射用水按照注射剂生产工艺制备所得,不含任何添加剂,属无菌状态[2]。2010年《药品生产质量管理规范》明确指出纯化水、注射用水的制备、贮存和分配是防止微生物的滋生的关键[3]。随着新版GMP的实施,各制药企业对制药用水风险控制更加关注,特别是水中微生物尤为重要。
皮氏罗尔斯顿菌是一种非发酵的革兰氏阴性杆菌,容易吸附在水系统的运输管道和储存罐中,大量繁殖并形成生物膜[4],且菌体较小可以通过0.02 μm过滤器,从而污染溶液,包括无菌注射用水、用纯净水制成的盐溶液、无菌药物溶液、冲洗液等[5-8],引发严重的医疗事故。因此寻找快速、准确的鉴定方法,对于制药企业对制药用水中皮氏罗尔斯顿菌的控制十分必要。
目前针对制药用水污染微生物的鉴定包括传统分离鉴定法、免疫学检测方法、分子生物学检测方法等[9]。传统微生物学方法中,皮氏罗尔斯顿菌生长缓慢,常需要培养72 h才可见圆形、凸起、透明小菌落,生化鉴定试验步骤繁琐、耗时较长。分子生物学方法中,对皮氏罗尔斯顿菌的鉴定主要是利用普通PCR技术,其它方法目前尚无报道。普通PCR技术操作复杂,特别是结果观察所采用的琼脂糖凝胶电泳法,需要耗时近60 min,大大增加了整个实验的操作时长。本实验采用PCR技术结合胶体金试纸条,只需要将PCR扩增产物滴加到胶体金试纸条上,从而实现5 min快速可视化结果观察,摆脱了琼脂糖凝胶电泳耗时较长的步骤,具有简单快速,成本较低的优点[10],更适合制药企业对污染菌的快速鉴定。本团队2021年对辽宁某生物科技有限公司制药用水采样,采用细菌16S rDNA通用引物扩增并测序,同一批污染菌标本鉴定出7株皮氏罗尔斯顿菌,以此对皮氏罗尔斯顿菌建立PCR-核酸试纸条快速检测方法并对此次污染进行溯源分析。
1 材料和方法
1.1 实验菌株
实验菌株来自制药企业制药用水中分离,采用细菌通用引物(上游引物27F:5'AGTTTGATCMTGG CTCAG3';下游引物1492R:5'GGTTACCTTGTTA CGACTT3')进行PCR扩增,由生工生物工程有限公司测序鉴定所得菌株。
1.2 试剂
LB培养基(OXOID)、磁珠保存管(青岛海博生物技术有限公司)、M-100 bp DNA Ladder、Taq PCR MasterMix(2X)(北京天根生化科技有限公司)、引物(上海生工生物有限公司)、生物素化牛血清白蛋白BSA-Biotin(北京达博诺科技有限公司)、链霉亲和素(北京金泰宏达生物科技有限公司)、兔抗FITC多克隆抗体(上海生工生物有限公司)、胶体金(上海易泽生物科技有限公司)、牛血清白蛋白(上海碧云天)、试纸条快速诊断整合方案(上海杰一生物技术有限公司)、其他试剂均为国产分析纯。
1.3 仪器
Thermo Fisher 高速离心机(Thermo)、NanoDrop One 微量核酸蛋白测定仪(Quawell)、金属浴(科路森)、漩涡混匀器(赛洛捷克)、多功能梯度PCR仪(东胜)、紫外凝胶成像分析仪(上海顾村电光仪器厂)、电泳仪(北京君意东方电泳设备有限公司)、恒温培养箱(上海博讯实验有限公司)、YXQ-LS-75S11立式蒸汽灭菌锅(上海博讯实业有限公司)。
1.4 煮沸法提取基因组
将制药用水污染菌样本进行编号,按编号分别进行分离培养,取分离培养后的实验菌株,用接种环取一环细菌于装有100 μL ddH2O的无菌离心管中,贴壁研磨至浑浊悬液。震荡混匀,将离心管放置金属浴中,100 ℃加热10 min,12 000×g水平离心10 min,弃沉淀,上清液为基因组DNA。
1.5 引物设计并标记
在NCBI上下载皮氏罗尔斯顿菌16S rDNA基因保守序列,通过DNAMAN比对分析,使用NCBI primer-BLAST 5.0设计一对特异性引物Ral-131,片段长度为131 bp(表1)。分别在引物的5’端标各标记异硫氰酸荧光素(FITC)与生物素(Biotin),由大连宝生物工程(大连)有限公司完成。
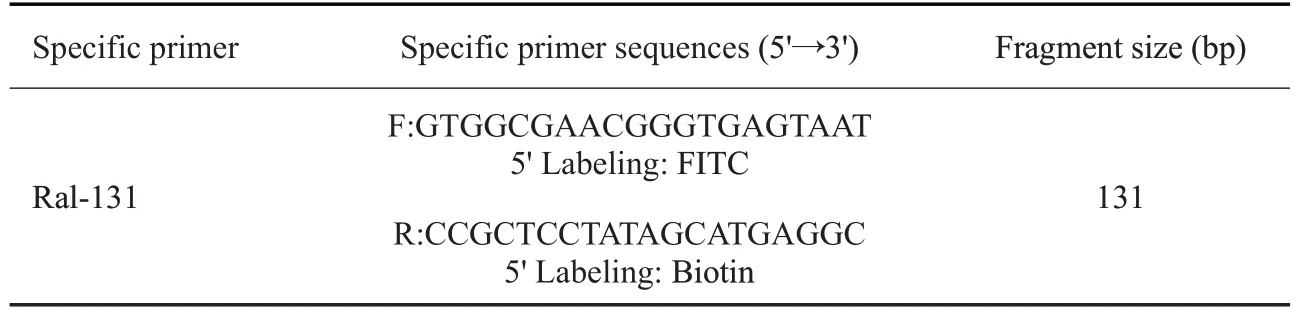
表1 皮氏罗尔斯顿菌特异性引物序列Table 1 Sequences of primers specific for Ralstonia pickettii
1.6 PCR反应体系
反应体系:PCR Master Mix(2×)12.5 μL,DNA模板(10 ng/μL)1 μL,特异性引物F、R(10 ng/μL)各0.5 μL,灭菌ddH2O补足至25 μL;反应条件:95 ℃预变性5 min;95 ℃变性30 s,59 ℃退火30 s,72 ℃延伸30 s,30 个循环;72 ℃充分延伸10 min,4 ℃低温保存。微量移液器取6 μL PCR产物加样于1.2%的琼脂糖凝胶中,85 V电泳50 min,紫外凝胶成像分析仪观察结果。
1.7 PCR产物克隆及测序、比对
将皮氏罗尔斯顿菌特异性DNA片段进行凝胶回收、纯化,测量回收DNA浓度;使用pGM-T连接试剂盒将回收产物DNA与pGM-T载体连接,连接后转入大肠杆菌感受态细胞DH5α,蓝白斑试验筛选阳性克隆;制备LB液体培养基增菌,进行16 h摇菌过夜;质粒小提试剂盒提取质粒DNA,以质粒DNA为模板进行扩增,PCR产物送至生工生物工程有限公司测序验证,分析比对结果。
1.8 PCR-核酸试纸条设计原理
以PCR 产物为检测样品,将8 μL PCR 产物与100 μL样品展开液混合,加入到试纸条的样品区,混合液由于毛细作用向前流动,当存在扩增产物时,扩增产物5’端标记的生物素首先与胶体金修饰的链霉亲和素相结合,液相到达检测线(T线)后,检测线上标记的兔抗FITC抗体会捕获扩增产物另一端修饰的异硫氰酸荧光素,形成复合物,从而使检测线显色。多余的胶体金颗粒会与质控线(C线)上的生物素化的牛血清白蛋白结合,使质控线显色。当不存在扩增产物时,T线上兔抗FITC抗体无法捕获到异硫氰酸荧光素,所以检测线不显色,而过量的胶体金修饰的链霉亲和素会与C线上的生物素化的牛血清白蛋白结合而显色(图1)。
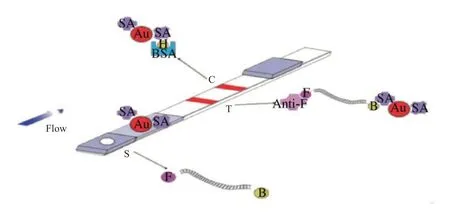
图1 胶体金核酸试纸条原理[11]Fig.1 Working mechanism of colloidal gold nucleic acid test strip[11].S: Sample area;T:Test line;C:Control line;AU:Colloidal aurum particle;SA:Streptavidin;B:Biotin;F:Fluorescein;Anti-F:Anti-fluorescein;BSA:Bovine serum albumin.
1.9 胶体金标记链霉亲和素最佳标记量的确定
将制得的胶体金用0.1 mol/L碳酸钾溶液调整pH值为7.0,分别取胶体金100µL于1.5 mL无菌EP管中。每个EP管中加入浓度为1 mg/mL待标记的链霉亲和素分别为2、2.5、3、3.5、4µL,震荡混匀10 min,对照管则不加,在胶体金链霉亲和素混合溶液中按0.5 mg/mL加入牛血清白蛋白,室温混合20 min后,将混合液12 000×g水平离心20 min,留取沉淀,将上清液继续按照上述方法离心,直至溶液无色,将沉淀悬浮于1/20~1/10初始胶体金体积的金胶缓冲液中悬浮使用,或于4 ℃保存[12-13]。以此确定链霉亲和素的最佳标记量。
1.10 抗体浓度的确定
将生物素化牛血清白蛋白和兔抗异硫氰酸荧光素抗体用磷酸盐缓冲液进行0.8、1.2、1.6、2 mg·mL-1稀释后分别划于硝酸纤维素膜上,形成质控线与检测线,37 ℃烘干,检测阳性PCR产物,根据显色结果确定两者的工作浓度。
1.11 试纸条的组装与检测
各部分按照相应的尺寸进行裁剪,将裁剪好的PVC底板、样品垫、制备完成的胶体金垫、标记好质控线与检测线的硝酸纤维素膜、吸水垫,按此顺序进行组装。用PCR产物进行检测,将PCR产物8µL和样品展开液100µL混合后加入样品垫上,5 min后观察结果。结果判定:仅当C、T 线同时出现红色条带为阳性结果;C线出现红色条带,T线没有出现红色条带为阴性;其他结果均为无效结果。
1.12 PCR-核酸试纸条原理的验证
核酸试纸条原理验证:将标记的引物和未标记的引物混合后进行PCR反应。具体方法:第1组为上下游均标记的引物;第2组为上游引物5’端标记的异硫氰酸荧光素与下游未标记的引物;第3组为上游未标记的引物与下游5’端标记的生物素的引物;第4组为均未标记的引物;预期仅有第1组结果显色。
1.13 PCR-核酸试纸条特异性评价
实验室对制药企业制药用水分离鉴定得到的不动杆菌,气单胞菌,假单胞菌、非脱羧勒克氏菌按照煮沸法提取基因组DNA,并稀释至10 ng/μL模板,进行核酸试纸条特异性进行评价,并与1.2%琼脂糖凝胶电泳结果进行比对。
1.14 PCR-核酸试纸条灵敏度评价
将皮氏罗尔斯顿菌DNA模板稀释7个浓度梯度,分别为101ng·μL-1、100ng·μL-1、10-1ng·μL-1、10-2ng·μL-1、10-3ng·μL-1、10-4ng·μL-1、10-5ng·μL-1,每份基因组取1 μL,按照上述反应进行核酸试纸条灵敏度评价,并与1.2%琼脂糖凝胶电泳结果进行比对。
1.15 PCR-核酸试纸条稳定性评价
按照优化的条件完成试纸条的组装,试剂的配制并保存。分别在3、6、9、12月进行核酸试纸条稳定性验证。PCR反应按照1.6节反应条件进行,并与1.2%琼脂糖凝胶电泳结果进行比对。
1.16 同源性分析并构建进化树
对本次注射用水所采集不同来源的7株皮氏罗尔斯顿菌使用细菌16S rDNA通用引物进行PCR扩增,并将测序结果使用MEGA 7.0进行16S rDNA基因序列同源性分析并对污染菌构建进化树,以此对此次污染菌进行溯源性分析。
2 结果
2.1 煮沸法提取基因组结果
使用NanoDrop One微量核酸蛋白测定仪对基因组测定,7株皮氏罗尔斯顿菌的A260/A280比值均介于1.8~2.0,纯度较好(表2)。经浓度0.8%琼脂糖凝胶DNA原液电泳结果显示,各泳道条带清晰明亮,此方法提取的基因组相对完整,符合PCR反应的要求。相比而言,试剂盒法提取基因组步骤较繁琐,耗时约90 min,而煮沸法整个过程仅需30 min。
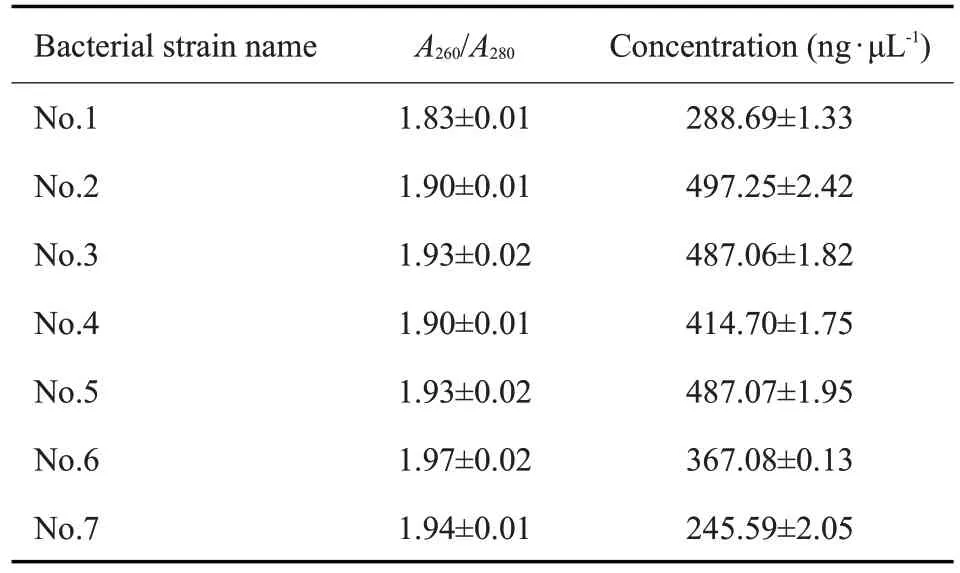
表2 皮氏罗尔斯顿菌DNA浓度、纯度结果Table 2 DNAconcentration and purity results of Ralstonia pickettii
2.2 PCR扩增结果
使用特异性引物对制药用水分离鉴定的7株皮氏罗尔斯顿菌进行PCR扩增,取6 μL PCR产物进行琼脂糖凝胶电泳,结果7株罗尔斯顿菌均在131 bp处有明亮单一条带(图2)。
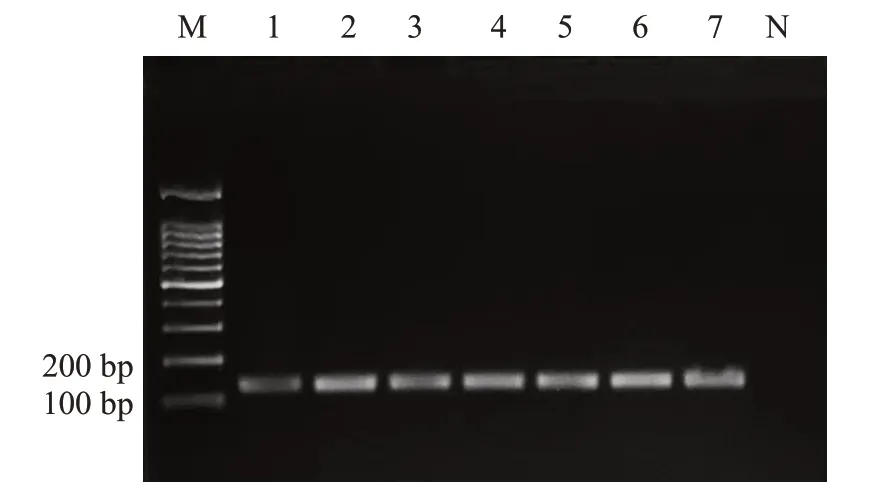
图2 PCR扩增电泳结果Fig.2 PCR amplification electrophoresis results.M-100 bp DNA ladder;Lanes 1-7: Ralstonia pickettii;N:Blank control.
2.3 PCR产物克隆及测序、比对结果
试剂盒提取质粒DNA经琼脂糖凝胶电泳结果显示:1、2泳道可见明亮清晰的目的条带(图3),证明目的基因成功转入质粒。PCR产物克隆后,测序分析结果显示(图4):经NCBI-BLAST比对,所测皮氏罗尔斯顿菌序列与GenBank中已登记的皮氏罗尔斯顿菌相似性为100%,证明所克隆片段为目的基因片段,可以作为阳性质粒。
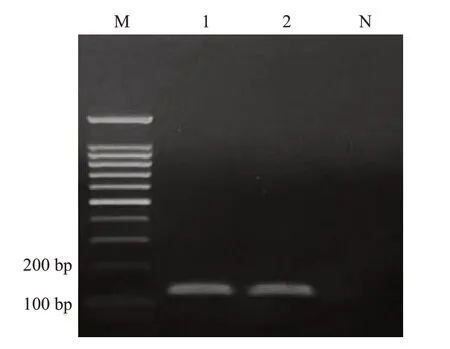
图3 克隆目的基因片段电泳结果Fig.3 Electrophoresis results of cloned target gene fragment.M: 100 bp DNA ladder;Lanes 1,2: Cloning target fragment of 1-2-Ralstonia pickettii;N:Blank control.

图4 皮氏罗尔斯顿菌目的基因序列比对结果Fig.4 Sequence alignment results of target genes of Ralstonia pickettii.
2.4 胶体金标记链霉亲和素的最佳标记量的确定
试验结果显示,1 mg/mL链霉亲和素的最适添加量为3.5 μL时,检测产物时显色度最好,颜色介于紫色和粉红色之间(图5),所以在后期组装试纸条时按3.5 μg进行标记。

图5 胶体金标记链霉亲和素的最佳标记量结果Fig.5 The optimal labeling amount of colloidal gold-labeled streptavidin.1:2µL,2:2.5µL,3:3µL,4:3.5µL,5:4.0µL.
2.5 抗体浓度的确定
试验结果显示,C线的生物素化牛血清白蛋白的最佳浓度为1.2 mg/mL时条带较清晰(图6)。T线上兔抗异硫氰酸荧光素最佳抗体浓度为2 mg/mL时,与阳性PCR产物反应,产生较为清晰的条带(图7)。
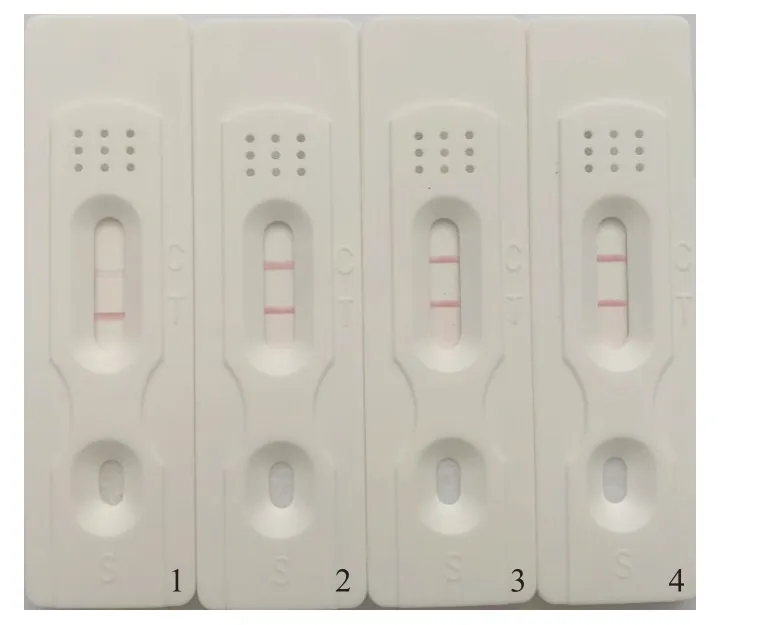
图6 生物素化牛血清白蛋白的浓度优化结果Fig.6 Concentration optimization results of biotinylated bovine serum albumin.1:0.8 mg·mL-1,2:1.2 mg·mL-1,3:1.6 mg·mL-1,4:2.0 mg·mL-1.

图7 兔抗异硫氰酸荧光素最佳抗体浓度优化结果Fig.7 Optimization results of rabbit optimal antibody concentration against fluorescein isothiocyanate.1:0.8 mg·mL-1,2:1.2 mg·mL-1,3:1.6 mg·mL-1,4:2.0 mg·mL-1.
2.6 核酸试纸条原理验证结果
本试验分别将5’端标记的引物和未标记的引物混合搭配后进行PCR 反应。只有标记引物的PCR 产物两端会形成异硫氰酸荧光素-生物素复合物,此时核酸胶体金试纸条才会发生显色反应。在保证同一PCR产物时,只有1号核酸胶体金试纸条为阳性结果(图8)。核酸试纸条原理验证试验成功。

图8 核酸试纸条原理验证结果Fig.8 Verification results of nucleic acid test strip principle.1:PCR products of both labeled upstream and downstream primers;2: PCR products of labeled upstream primers and unlabeled downstream primers;3: PCR products of unlabeled upstream primers and labeled downstream primers;4: Both upstream and downstream primers that are not labeled PCR product.
2.7 PCR-核酸试纸条特异性评价
对本次采样的7株皮氏罗尔斯顿菌与实验室保存的制药用水分离鉴定的不动杆菌,气单胞菌,假单胞菌、非脱羧勒克氏菌进行实验特异性的评价。电泳结果显示,7株不同来源的罗尔斯顿菌均在131 bp处扩增出单一明亮目的片段,其它菌株未见条带(图9)。PCR-核酸试纸条的结果显示,仅有皮氏罗尔斯顿菌为阳性结果,其它菌株及空白对照只有质控线的出现红色条带,为阴性结果(图10)。PCR电泳结果与PCR-核酸试纸条结果一致。
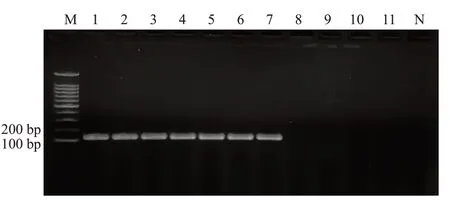
图9 引物特异性验证Fig.9 Primer specificity verification.M-100 bp DNA ladder;1-7: Ralstonia pickettii;8: Leclercia adecarboxylata;9:Acinetobacter calcoaceticus;10: Pseudomonas;11: Aeromonas;N-blank control.

图10 核酸试纸条特异性验证Fig.10 Specificity verification of nucleic acid test strip.1-7:Ralstonia pickettii;8:Leclercia adecarboxylata;9:Acinetobacter calcoaceticus;10:Pseudomonas;11:Aeromonas;N:blank control.
2.8 PCR-核酸试纸条灵敏度评价
采用无菌ddH2O将皮氏罗尔斯顿菌DNA模板稀释7个浓度梯度,对实验进行灵敏度评价。电泳结果显示,在10-2ng·μL-1时仍可见较浅目的条带,证明引物灵敏度良好,最低检测限为10-2ng·μL-1(图11)。PCR-核酸试纸条的结果显示,在稀释到10-5ng·μL-1仍可见C线和T线同时出现两条清晰的红色条带,为阳性结果,所以试纸条的最低检测限为10-5ng·μL-1(图12)。PCR-核酸试纸条的灵敏度较普通PCR灵敏性高1000倍。

图11 引物灵敏度验证Fig.11 Primer sensitivity test.M:100 bp DNA Ladder;1:101 ng·µL-1;2:100 ng·µL-1;3:10-1 ng·µL-1;4:10-2 ng·µL-1;5:10-3 ng·µL-1;6:10-4 ng·µL-1;7:10-5 ng·µL-1;N:Blank control.
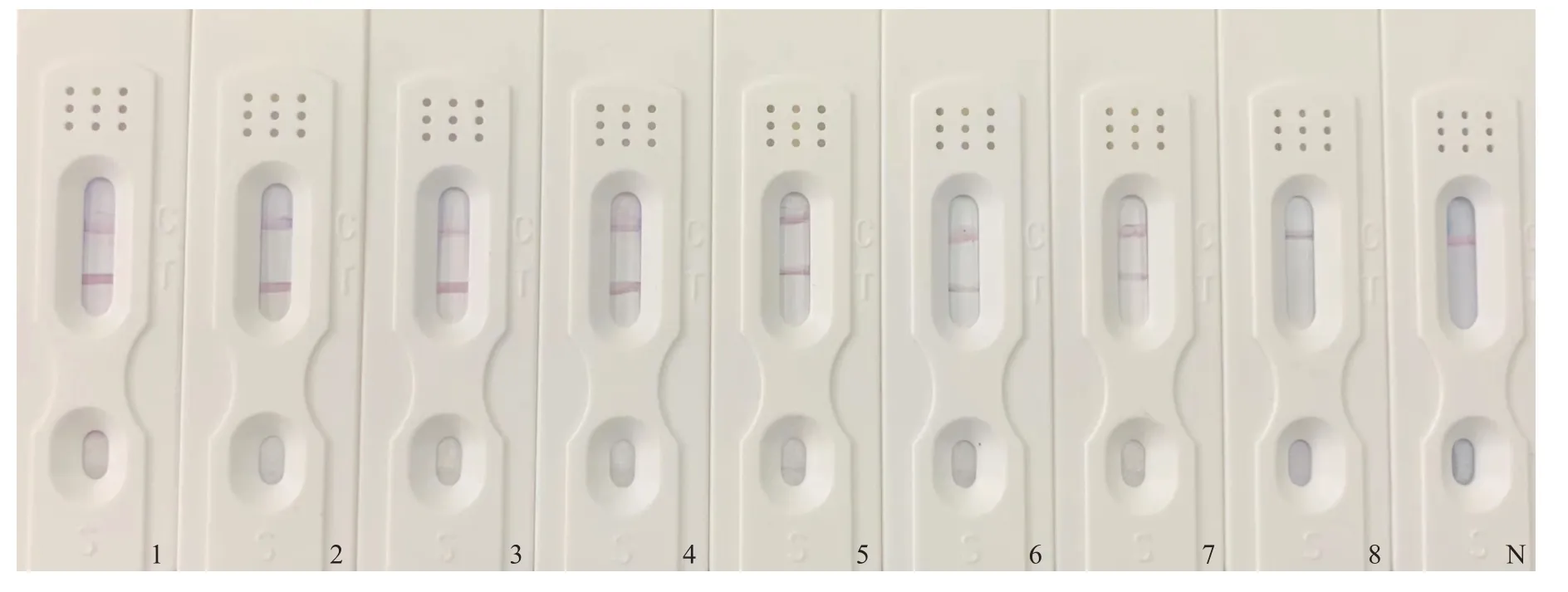
图12 核酸试纸条灵敏度验证Fig.12 Sensitivity test of nucleic acid test strip.M:100 bp DNALadder;1:101 ng·µL-1;2:100 ng·µL-1;3:10-1 ng·µL-1;4:10-2ng·µL-1;5:10-3 ng·µL-1;6:10-4 ng·µL-1;7:10-5 ng·µL-1;8:10-6 ng·µL-1;N:Blank control.
2.9 PCR-核酸试纸条稳定性评价
按照优化的反应条件组装核酸试纸条,将保存的核酸试纸条在第3、6、9、12月取出进行稳定性验证,结果显示,与阳性对照相比较,阳性PCR产物检测结果一致,只有皮氏罗尔斯顿菌出现阳性结果即C线、T线都出现红色条带,空白对照为阴性结果即只C线出现1条红色条带(图13)。

图13 试纸条3、6、9、12月稳定性验证Fig.13 Stability verification of test strip at 3(A),6(B),9(C)and 12 months(D).1:Positive control of Ralstonia pickettii;2:Ralstonia pickettii;3:Blank control.
2.10 构建进化树进行溯源性分析结果
对此次注射用水采集鉴定的7株皮氏罗尔斯顿菌,分别来自纯化水区、注射用水区、细胞反应器与不同规格的细胞培养基,使用MEGA 7.0进行16Sr DNA基因序列同源性分析并对污染菌构建进化树(图14)。根据发育树值显示,纯化水区、注射用水区、细胞培养基与细胞培养反应器菌株亲缘关系较近,考虑存在交叉污染,应对制药用水区进行监测。
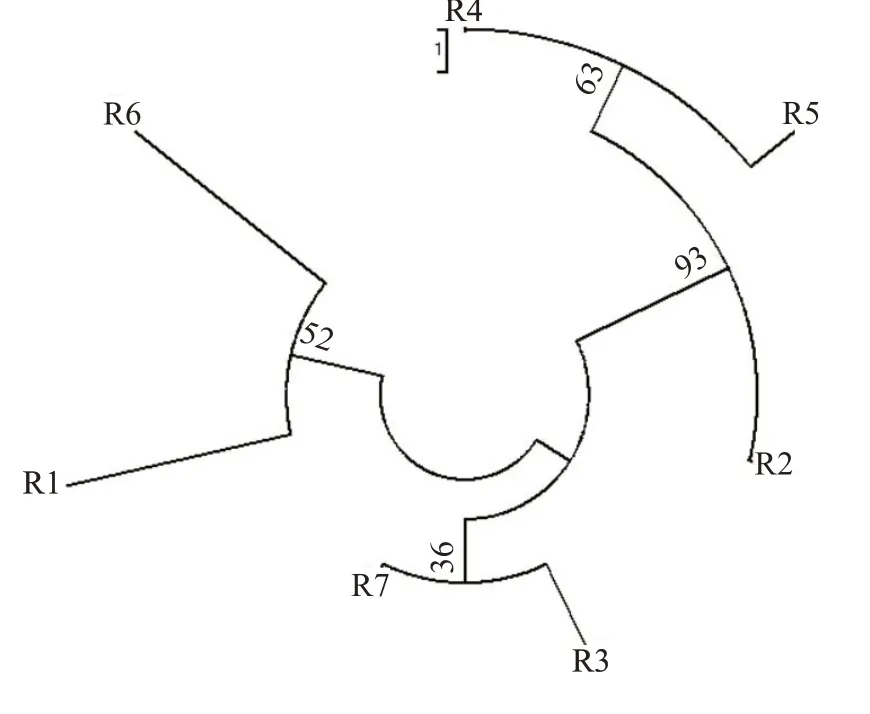
图14 7株皮氏罗尔斯顿菌16S rDNA构建系统发育树Fig.14 Phylogenetic tree of 16S rDNA construction of 7 strains of Ralstonia pickettii.R1: Purified water area;R2:Injection water area;R3: 300 L bacterial culture medium;R4: 200 L bacterial culture medium;R5: 200 L bacterial culture medium;R6:300 L bacterial culture medium;R7:Medium reactor.
3 讨论
皮氏罗尔斯顿菌曾被认为是一种毒性很低,常常被忽略的致病菌,但在医院环境中,它们已被证明会引起严重的医院感染[14-19],如脑膜炎、肺炎、血流感染等。同时在治疗方面这种细菌是很顽固的,它对多种不同类型的抗生素产生耐药性[20]。因此建立快速鉴定制药用水中皮氏罗尔斯顿菌的方法,保障用药安全尤为重要。
随着分子生物学技术的发展,采用PCR技术鉴定微生物的方法逐渐成熟且多元化。有研究针对高风险注射剂生产企业的制药用水系统建立微生物菌库,纯化水系统和注射用水系统中通过PCR扩增及测序技术鉴定出多株罗尔斯顿菌[21],但没有针对这类常见污染菌建立快速检测的方法。Ryan 等[22]使用多重PCR 技术、RAPD-PCR和BOX-PCR技术对皮氏罗尔斯顿菌进行鉴定,但此方法需要操作人员具有一定的专业知识,步骤繁琐,使用琼脂糖凝胶电泳法观察结果,耗时较长。而相比PCR-核酸试纸条技术就无须耗时较长的电泳技术,它基于PCR技术设计特异性引物,并将引物进行双标记,充分体现了PCR技术的高灵敏度与特异性,产物中的FITC与胶体金试纸条上相对应的抗体结合形成检测线,生物素-胶体金修饰的链霉亲和素与生物素化的牛血清白蛋白特异性结合形成质控线,整体反应进行优化,既可以使得整个鉴定过程可在2 h内完成,又可以避免生物素过饱和而导致的阳性产物漏检情况的发生。这种可靠的检测技术可以为质检人员提供现场快速且操作简单的检测手段,大大缩短污染菌的鉴定周期。在节约成本方面,核酸胶体金试纸条相比免疫胶体金试纸条可以在购买昂贵抗体方面减少资金花费,从而降低胶体金试纸条的成本,更适合基层制药企业使用。
本实验以制药用水中分离得到的7株皮氏罗尔斯顿菌为研究对象,将PCR技术与胶体金免疫层析法相结合,建立PCR-核酸试纸条技术快速检测皮氏罗尔斯顿菌。因煮沸法较试剂盒法提取基因组在耗时上更有优势,且利用煮沸法提取基因组DNA,浓度、纯度较好、操作简单、可快速获得,已通过克隆转化得到阳性对照。检测时只需要将阳性PCR产物8 μL与100 μL样品展开液混合后滴加到样品垫上,5 min 后即可观察结果。实验特异性结果与琼脂糖凝胶电泳结果一致,灵敏度较琼脂糖凝胶电泳高1000倍,在试纸条3、6、9、12月分别检测稳定性,结果表明稳定性较好,因此核酸试纸条技术也被应用到其它多个领域[23-25]。本实验研制的核酸胶体金试纸条比起传统微生物学方法、普通PCR技术与免疫胶体金技术等有一定的优越性,但对于一些基层企业的分子生物学实验室没有PCR仪等先进仪器,不能做到及时且快速的检测,为了解决这些问题,更好的服务于基层企业,本实验后续将考虑使用不需要复杂仪器即可完成反应的等温扩增技术,即LAMP[26]、RPA[27]及PSR[28]等技术,它们与试纸条技术相结合必将成为未来检测技术发展的新方向,从而更好的为制药企业建立快速检测污染菌提供更多地技术支持。
