人血清新miRNA分子的预测及表达分析*
2022-01-24李晓辉林发全曾麒燕
李晓辉,刘 奕,林发全,肖 毅,侯 伟,熊 灏,曾麒燕,5,6△
(1.广西医科大学生物化学与分子生物学教研室,南宁 530021;2.广西医科大学第一附属医院检验科,南宁 530021;3.广西医科大学生命科学研究院地中海贫血防治重点实验室,南宁 530021;4.广西卫生职业技术学院医技系,南宁 530021;5.广西高校生物分子医学研究重点实验室,南宁 530021;6.长寿与老年相关疾病教育部重点实验室,南宁 530021)
外泌体是细胞释放的一类脂质双分子膜包裹的直径为30~150 nm 的囊泡,其中富含蛋白质、核酸和脂质[1]。外泌体通过与靶细胞融合,传输物质并传递信号,从而改变细胞生理状态[2]。越来越多的研究表明,外泌体在细胞发育、免疫防御、肿瘤发生和神经退行性疾病等方面发挥重要调控作用[3]。MicroRNAs(miRNAs)是一种长度大约为22 个核苷酸的内源性单链非编码小RNA。成熟miRNA主要通过其种子序列(即5’端第2~8位核苷酸)与靶基因mRNA 3’端非翻译区(3’-UTR)特异性碱基配对,从而以切割、降解mRNA 或抑制mRNA 翻译等形式调节基因表达[4]。miRNA 在血清中表达稳定,已成为肿瘤诊治及预后评估的理想生物标志物。血液中循环miRNA有3种形式:游离miRNA、Argonaute 家族蛋白结合的miRNA 和外泌体miRNA,其中以外泌体携带为主[5]。
本研究首先分离4例正常人和4例乳腺癌患者血清外泌体,然后进行Illumina HiSeq SE50 高通量miRNA 测序,滤掉已知miRNA 后,利用Bowtie 2、MIREAP、miRBase 和RNAfold 等生物信息学手段筛选新miRNA 序列,并对其长度分布、二级结构进行分析,并进一步分析其在正常人和乳腺癌患者血清中的表达水平。最后通过miRWalk 2.0、GEPIA和UALCAN 等数据库预测新miRNA的靶基因及其在乳腺癌中临床意义,以期为肿瘤诊治和预后评估提供依据。
1 材料与方法
1.1 实验材料
2018 年9月分别收集4例正常女性和4例未接受治疗的乳腺癌术前女性患者(来自广西医科大学肿瘤医院)的血清样本,用于外泌体提取。另于2020年10月至2021年1月分别收集18 例正常女性和29 例未接受治疗的乳腺癌术前女性患者(均来自广西医科大学第一附属医院)的血清样本,用于对候选新miRNAs表达水平的分析。本实验获得广西医科大学伦理委员会批准,并获得患者及志愿者的知情同意。
1.2 主要试剂
miRNeasy Serum/Plasma Kit(Cat.No.217184)、QIAzol Lysis Reagent(Mat.No.1023537)购自德国Qiagen 公司;Mir-X miRNA First Strand Synthesis Kit(Cat#638315)、TB Green Premix Ex TaqTMⅡKit(Cat # RR047A)购自日本TaKaRa 公司;新miRNA上游引物,hsa-miR-23a-3p 和U6 上游引物、通用下游引物(Cat#638315)均由上海生工生物工程有限公司提供。
1.3 实验方法
1.3.1 外泌体的提取和miRNA 测序 根据文献[6],利用超速离心法分离血清外泌体,于透射电镜下观察其形态,同时检测其粒径大小和标志分子,并进一步通过Illumina HiSeq SE50 平台(武汉承启医学科技有限公司)对外泌体进行高通量miRNA测序。
1.3.2 新miRNA 分子预测 对测序所得原始数据去除低质量序列、去接头序列后得到clean reads。利用短序列比对工具bowtie 2[7],将clean reads 与人全基因组数据库(GRCh38.P13)比对,允许错配为0,将比对上的序列与GenBank 数据库和Rfam 数据库比对,去掉已知miRNA、rRNAetc、repeat、外显子和内含子等序列,得到新miRNA 候选序列。利用RNAfold 软件预测新miRNA 前体序列的二级结构及其最小自由能;并进一步用MIREAP 软件[8]分析候选序列,参数设置[9]如下:(1)miRNA 序列长度18~26 个核苷酸;(2)miRNA 参考序列长度20~24个核苷酸;(3)miRNA 在参考序列上最大拷贝数为20;(4)miRNA 前体允许的最大自由能为-18 kcal/mol;(5)miRNA和miRNA*之间空格数为5~35;(6)miRNA 和miRNA*之间最大凸环为4;(7)miRNA和miRNA*之间最小配对数为14;(8)侧翼序列长度为100 nt。
1.3.3 实时荧光定量PCR(RT-qPCR)分析新miRNA在血清中的表达水平 用miRNeasy Serum试剂盒提取血清miRNA,然后用Mir-X miRNA First Strand Synthesis Kit对miRNA进行逆转录。采用加尾法对其中两条新miRNA 进行RT-qPCR,同时,以血清中稳定表达的hsa-miR-23a-3p 作为阳性对照,以U6 作为内参,用2-△△Ct计算miRNAs 的相对表达量。反应条件如下:预变性95 ℃30 s;95 ℃15 s,55 ℃30 s,72 ℃30 s,共40个循环。引物序列如下:miRNA-A 上游:5’-CCTTGTCCATAAACCTGGATCGAGG-3’;miRNA-B 上游:5’-TATATATAGGGGCCTCGAGCCGC-3’;hsa-miR-23a-3p 上游:5’-CCGATCACATTGCCAGGGATTTCC-3’;U6 上游:5’-TAGCAGCACGTAAATATTGGCG-3’;所有通用下游引物均由TaKaRa 公司提供。
1.3.4 新miRNA 分子靶基因预测及其在乳腺癌中表达 用miRBase 数据库查找与新miRNA 种子序列完全一致的已知miRNA,再用miRWalk 2.0 数据库预测该已知miRNA 的靶基因,最后通过Needle软件比对候选靶基因3’-UTR 与新miRNA 的种子序列的匹配程度,确定其是否为新miRNA的候选靶基因,并进一步利用GEPIA 和UALCAN 数据库预测靶基因在乳腺癌中的表达水平及其与分子分型和组织学分型的关系。
1.3.5 统计学方法 采用SPSS 25.0软件进行数据分析。计量资料以均数±标准差()表示,组间比较采用独立样本t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 外泌体的鉴定
透射电镜下可观察到外泌体呈圆盘状,膜结构完整、轮廓清晰;粒径分析显示粒径范围20~200 nm,平均38.17 nm,主峰71.26 nm;高表达CD63、CD81、CD9、TSG101及HSC70等分子标志物,表明提取的外泌体质量良好,详细鉴定结果见课题组的前期文献报道[6]。
2.2 新miRNA筛选
经miRNA测序,从正常组和乳腺癌患者样本中分别获得16 394 816 和14 535 792 条reads,经处理冗余数据后分别得到5 173 401 和4 695 648 条clean reads。用短序列比对工具Bowtie 2 将clean reads与人全基因组数据库序列进行比对后,正常组和乳腺癌患者组中比对上的reads 数目分别为1 874 074 和1 980 374 条。去掉已知的miRNA、rRNAetc、外显子、内含子和重复序列,正常组和乳腺癌患者组分别剩下130 699 和128 889 条reads。为了寻找正常组和患者组间差异的新miRNA分子,将上述两组reads 进行比较,并根据miRNA 预测原则筛选出7 条可能存在差异表达的新miRNA 候选序列,分别命名为miRNA-A、miRNA-B、miRNA-C、miRNA-D、miRNA-E、miRNA-F和miRNA-G(表1),7 条新miRNA 前体分子的二级结构(图1),其成熟序列均来自茎—环前体的一个臂且拥有较低的自由能,符合miRNA预测原则。
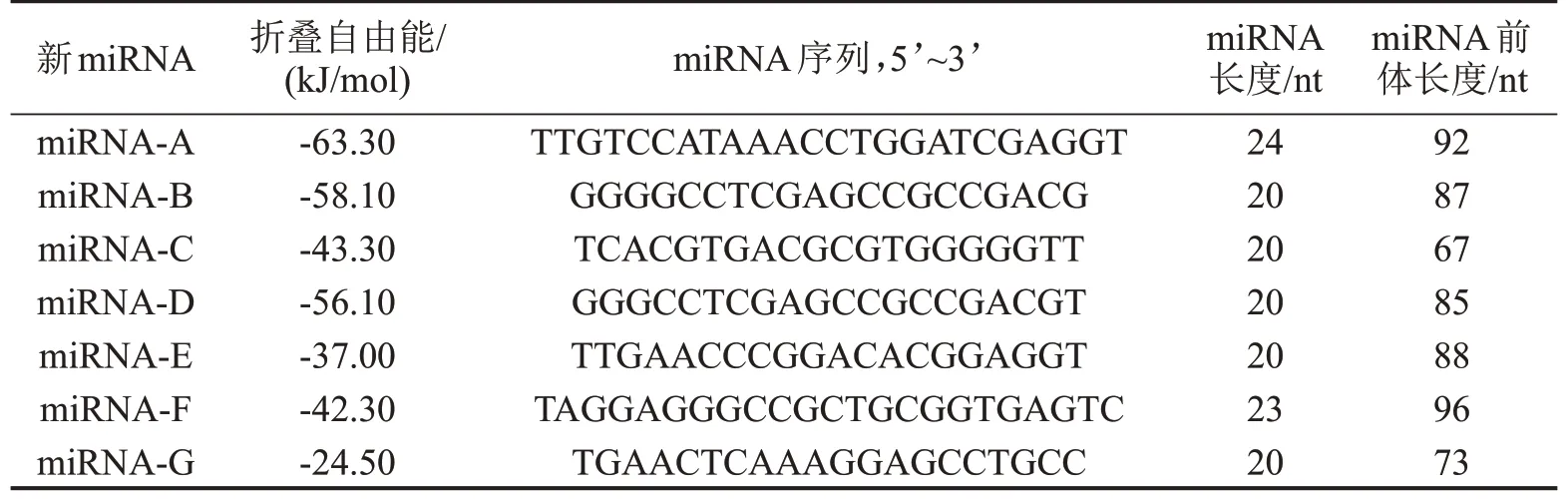
表1 7个候选新miRNA分子

图1 候选新miRNA前体分子的二级结构预测
2.3 新miRNA在血清中的表达水平分析
用RT-qPCR 分析miRNA-A、miRNA-B 在血清中的表达水平,并以hsa-miR-23a-3p 作为阳性对照。结果显示,miRNA-A 和miRNA-B 在正常组和乳腺癌组血清中均表达,但无显著差异(P>0.05)(图2),而hsa-miR-23a-3p水平在乳腺癌组中显著下调(t=-2.767,P=0.013)(图2),与文献[10]报道一致。
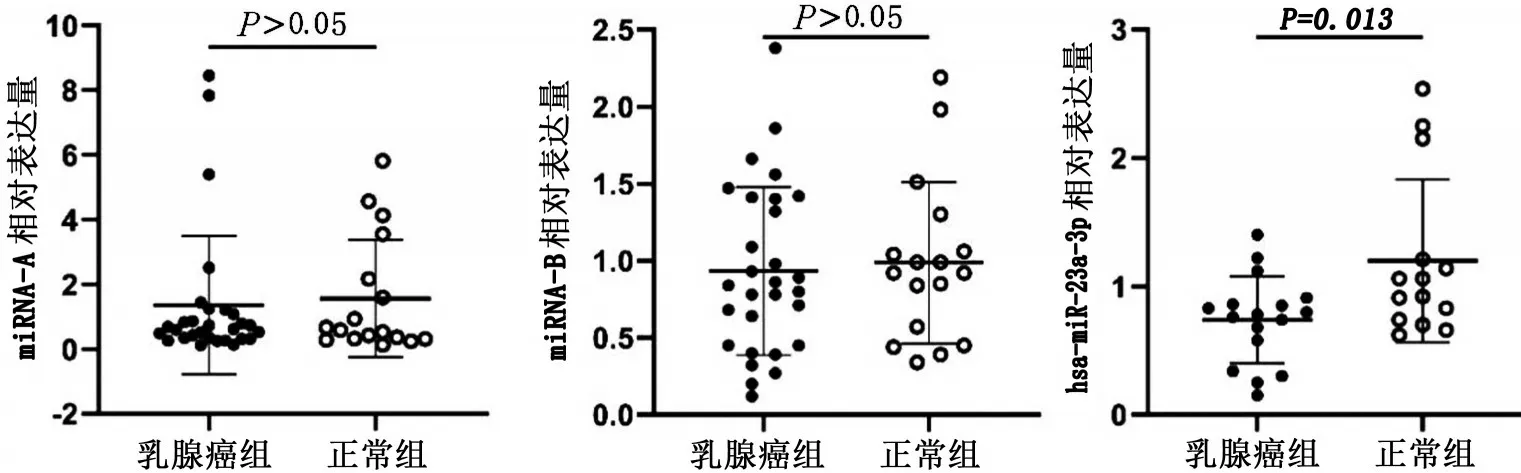
图2 miRNA-A、miRNA-B和hsa-miR-23a-3p在血清中的表达水平分析
2.4 新miRNA靶基因预测
种子序列是动物miRNA 识别并结合靶基因的关键功能元件,其突变可导致miRNA 功能的改变[11]。通过miRBase 数据库,发现hsa-miR-3912-5p的种子序列与miRNA-A 的种子序列完全一致。利用miRWalk 2.0数据库预测hsa-miR-3912-5p的靶基因,选取同时出现在4个以上miRNA靶基因预测平台且排序前400的靶基因,验证其与新miRNA-A的匹配程度,最后发现4个与miRNA-A的种子序列完全匹配,并且整体序列匹配率较高靶基因,分别为CLK1(CDC like kinase 1)、CLK4(CDC like kinase 4)、GNG2(G protein subunit gamma 2)和NPR2(natriuretic peptide receptor 2)。miRNA-A 种子序列与靶基因匹配情况,见图3。
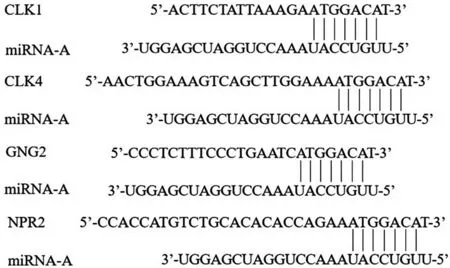
图3 预测靶基因与miRNA-A的匹配情况
2.5 新miRNA靶基因在乳腺癌中的表达分析
通过GEPIA 和UALCAN 数据库分析发现CLK1、CLK4、GNG2 和NPR2 在乳腺癌组织中均显著下调(均P<0.05)(图4)。根据分子表达谱不同,乳腺癌可分为Luminal型、HER2阳性及三阴性乳腺癌;根据是否浸润可分为非浸润癌(原位癌)及浸润性癌,后者又分为浸润性导管癌、浸润性小叶癌及特殊类型癌。进一步分析发现,CLK1、CLK4、GNG2 和NPR2 在HER2 阳性患者中的表达水平均显著低于Luminal型患者中的水平(均P<0.05)(图5A);浸润性导管癌(IDC)患者中的表达水平显著低于浸润性小叶癌(ILC)患者(P<0.05)(图5B)。
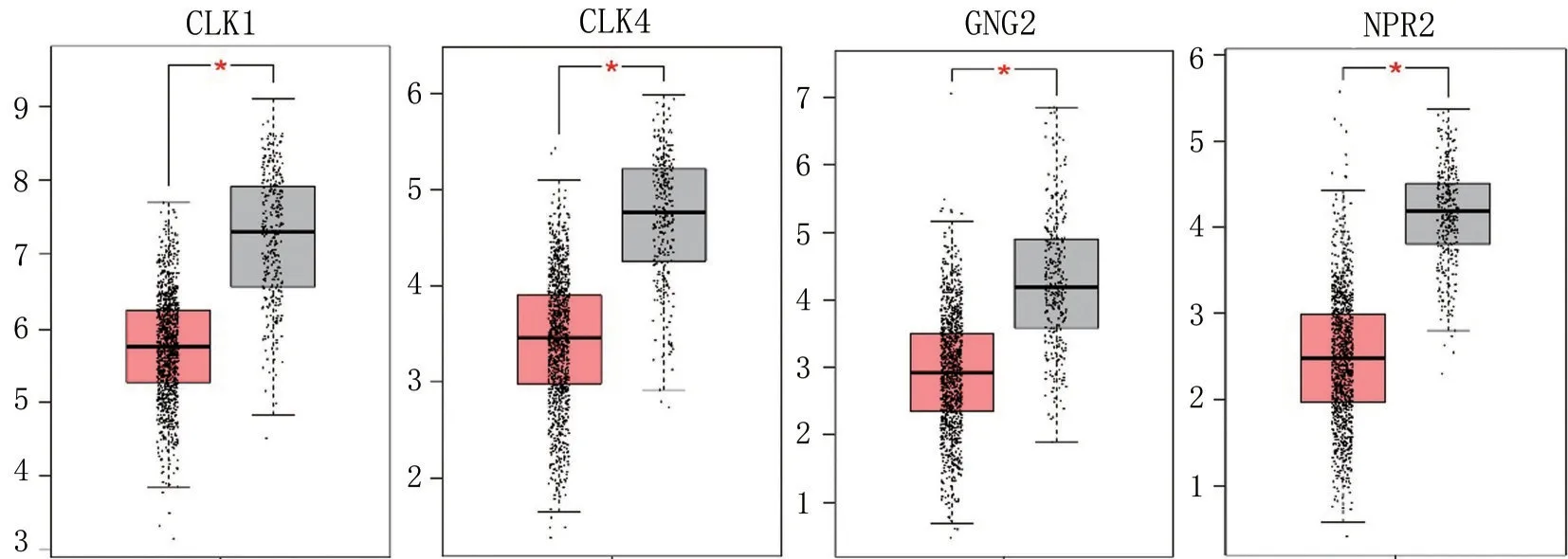
图4 靶基因CLK1、CLK4、GNG2和NPR2在乳腺癌组织中的表达分析
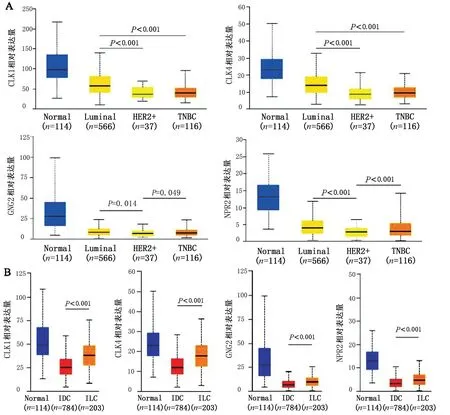
图5 靶基因CLK1、CLK4、GNG2和NPR2在不同分型乳腺癌中的表达水平
3 讨论
miRNA在基因调控中发挥重要作用,虽然其功能尚未完全明了,但已有研究表明miRNA与乳腺癌发生发展密切相关,如miR-155 通过靶向TRF1 基因,增加染色体端粒不稳定性,从而促进乳腺癌发生[12]。血清miRNA因其高度的组织特异性、稳定性及其在疾病发生发展中水平变化而被认为是乳腺癌诊断和预后评估的生物标志物。如血浆miR-21水平显著高于健康人和乳腺良性肿瘤患者,被认为是乳腺癌诊断的标志物[13]。miRNA 联合分析可进一步提高诊断的特异性和准确度,如miR-139-5p、miR-10b-5p、miR-486-5p、miR-455-3p、miR-107、miR-146b-5p、miR-324-5p 和miR-20a-5p 8 个miRNA联合预测模型可为三阴性乳腺癌患者的复发风险及预后进行评估[14]。
高通量测序技术被广泛用于动植物小RNA 的测序鉴定[15]。本研究通过Illumina HiSeq SE50对正常人和乳腺癌患者血清外泌体进行miRNA测序,得到2 347个已知miRNA,与正常人相比,在乳腺癌患者中有67个miRNA表达上调,56个miRNA表达下调。其中|log(Fold Change)|>9的显著上调的miRNA 有miR-451b、miR-27b-5p、miR-320b、miR-433-5p 和miR-299-3p 等,显著下调的有miR-9-5p、miR-874-5p 和miR-511-5p。研究已证实,miR-451 在乳腺癌患者血清中高表达[16],miR-9 在乳腺癌患者血清中低表达[17],这与本课题组的测序结果一致。而miR-27b-5p、miR-320b、miR-320c、miR-433-5p、miR-299-3p、miR-874-5p和miR-511-5p 在乳腺癌血清中的表达水平有待进一步验证。此外,我们结合生物信息学方法筛选出7个候选新miRNA 分子,序列长度在18~25 nt,为典型的Dicer 酶切产物,并且这些候选miRNA 分子的二级结构均为典型的不完全配对茎环结构,且具有较小的自由能,符合miRNA 的特征。我们选取其中2 个候选miRNA(miRNA-A、miRNA-B)分析,发现它们在正常组和乳腺癌组血清中均有表达,但两组比较差异无统计学意义(P>0.05),可能与样本量较少有关。至于它们在其他疾病中的表达水平与正常人是否有差异,有待进一步研究。
本研究通过miRWalk 2.0 数据库预测得到miRNA-A 的4个靶基因,分别为CLK1、CLK4、GNG2和NPR2。研究表明,CLK1 和CLK4 定位于细胞中间体,与细胞分裂密切关系[18],过表达GNG2能抑制乳腺癌细胞增殖,延长细胞周期进程[19],NPR2 与雌激素、雄激素、促卵泡素和黄体生成素等性激素分泌有密切关系[20],而机体激素水平紊乱也是乳腺癌发病的因素之一,因此,我们推测CLK1、CLK4、GNG2和NPR2可能通过调节细胞周期进程以及激素水平而影响乳腺癌的发生发展。通过GEPIA 和UALCAN 数据库分析发现这4 个靶基因在乳腺癌组织中均显著下调,进一步分析发现4 个靶基因在HER2 阳性患者中的表达水平均显著低于Luminal型患者,同时,在IDC 患者中的表达水平也均显著低于ILC患者。研究表明,HER2+乳腺癌患者的临床表现和病理性反应的程度明显强于Luminal HER2-患者,并且其预后也较差[21]。另一项研究表明,IDC 患者的总体生存率明显较ILC 患者低[22]。因此,本研究 结果提示CLK1、CLK4、GNG2 和NPR2这4个靶基因与乳腺癌疾病的严重程度相关,miRNA-A 可能通过调节这4 个靶基因的表达来调控细胞周期进程和机体激素水平,从而促进乳腺癌的发生发展,我们后续将继续深入研究miRNA-A与乳腺癌的关系。
综上,本研究通过高通量测序结合生物信息学方法筛选得到新的候选miRNAs,进一步分析了其中两个新miRNA 在血清中的表达水平,并预测其靶基因,新miRNA 的生物学功能及其与疾病的关系有待进一步深入研究。
