土壤铵氮在热活化过硫酸盐氧化过程中的转化
2022-01-21杨培增岳泓伸季跃飞陆隽鹤
杨培增,岳泓伸,季跃飞,陆隽鹤
土壤铵氮在热活化过硫酸盐氧化过程中的转化
杨培增,岳泓伸,季跃飞,陆隽鹤*
(南京农业大学资源与环境科学学院,江苏 南京 210095)
采用来自江苏和河北,具有不同土壤有机质含量和NH4+浓度的土壤样本,系统地研究了NH4+在热活化过硫酸盐(PS)氧化过程中的转化和归趋,考察了反应时间、PS浓度和外加NH4+对硝基副产物生成的影响.结果表明,土壤中的NH4+能够转化成3-硝基酚、4-硝基酚、2-羟基-5-硝基苯甲酸、4-羟基-3-硝基苯甲酸、2,4-二硝基酚等副产物,它们的生成量随着反应的进行先增加后降低.增大PS浓度可促进硝基副产物的生成.当PS浓度为30mmol/kg,反应12h后一硝基酚和一硝基羟基苯甲酸的生成量达到最大.然而随着PS浓度进一步增大,硝基副产物发生降解.硫酸根自由基(SO4•-)在硝化过程中起到了关键作用,它能将NH4+氧化生成氨基自由基(•NH2),随后经过一系列自由基链式反应生成二氧化氮自由基(NO2•).同时,SO4•-进攻土壤有机质中的酚结构单元,使其氧化生成苯氧自由基,苯氧自由基进一步与NO2•结合生成硝基副产物.天然有机质(NOM)在环境中无处不在,NH4+在环境中也普遍存在,PS用于土壤和地下水污染修复时生成硝基副产物很可能是一个普遍现象.
铵氮;过硫酸盐;土壤有机质;硫酸根自由基;二氧化氮自由基;硝基副产物
过硫酸盐(PS)高级氧化技术是近年来的研究热点[1-2].PS在常温下较稳定,很难氧化有机污染物[1].一般情况下,PS可以通过加热、紫外光照、过渡金属离子等方式活化产生硫酸根自由基(SO4×-)[1-4]. SO4×-的氧化还原电位(0=2.5~3.1V)和羟基自由基(×OH)相当(0=1.8~2.7V),但比×OH稳定;且SO4×-降解有机污染物时受pH值的影响小,有利于在更广泛的环境条件中使用[5-6].此外,SO4×-的寿命比×OH长,这大大提高了有机污染物与SO4×-的接触机会,从而有利于有机污染物的降解与矿化[5].基于以上优点,过硫酸盐高级氧化技术在近些年迅速发展并被广泛应用于有机污染物的去除[7-14].例如,Waldermer等[11]采用热活化PS去除地下水中的氯代乙烯,结果表明,当温度设定为60℃,PS浓度为0.45mmol/L,反应1h后,氯代乙烯的去除率接近100%.刘小宁[12]研究发现,热活化PS降解对氯苯酚的反应速率随温度升高而升高,当PS浓度为40mmol/L,60℃反应2h后,对氯苯酚去除率达到100%.Liu等[13]研究发现,当PS为20mmol/kg,温度从30℃上升至60℃,反应1h后,土壤中布洛芬的去除率从10%上升至100%.这些研究均反映出热活化PS(式1)降解有机污染物的高效性.与其他活化方法相比,热活化还有如下优点:首先,热活化操作简便,可控性强;其次,这种方法可以避免引入其他的离子,相对干净.此外,热活化受pH值的影响小,可以在各种pH值条件下使用.
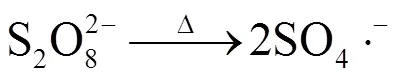
然而,越来越多的研究发现过硫酸盐高级氧化过程中有害副产物的生成.例如,SO4•-可以与卤素离子反应生成游离卤以及卤自由基,这些活性卤物质会进攻酚类底物生成有毒的卤代副产物,如卤仿和卤乙酸等[15-18].近来研究发现,SO4×-也能与土壤或地下水环境中广泛存在的亚硝酸盐(NO2-)发生反应,生成二氧化氮自由基(NO2×)(式2)[19-21].NO2×是一种较为温和的氧化剂,还原电位0为1.03V,在天然有机质(NOM)存在下,NO2×能够和NOM反应,生成硝基副产物[19-21].其中,NOM大分子中的酚结构单元为主要的活性位点.SO4×-通过电子转移能够将酚结构单元氧化成苯氧自由基,苯氧自由基进一步与NO2×结合生成硝基副产物[22-23].
=8.8×108mol/(L×s) (2)
铵(NH4+)是水和土壤中含量最为丰富的无机氮.铵通常作为肥料被大量释放到环境中,是造成地表水的富营养化的主要因素之一.土壤中NH4+-N含量从几mg/kg到几百mg/kg不等[24].研究表明,NH4+可以被×OH氧化形成氨基自由基(×NH2,0=2.3V),后者可以与氧气快速结合生成过氧氨基自由基(NH2OO×)[25-32],NH2OO×经过重排后分解生成氮氧自由基(NO×,0=0.39V)[25-26].NO×与×OH或氧气反应,生成NO2-或NO2×[33-36](式(3)~(8)).理论上,NH4+也可以被SO4×-氧化为硝酸盐(NO3-),但尚未有相关研究的报道[25].由于SO4×-与×OH氧化能力相近,可以推测NH4+在SO4×-氧化过程中具有类似的转化途径.一旦NH4+能够转化生成NO2×,在有机质存在的条件下,NO2×很可能与有机质反应生成硝基副产物.

=1.4×107mol/(L×s) (3)
=3.0×108mol/(L×s) (4)
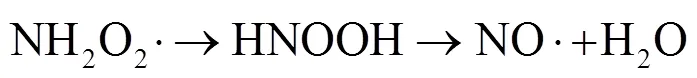
=1.1×109mol/(L×s) (5)
=1.7×1010mol/(L×s) (6)

=1.6×106mol/(L×s) (7)
=1.0×1010mol/(L×s) (8)
因此,本研究采用热活化过硫酸盐处理土壤,探究该过程中NH4+的转化及硝基副产物的生成,考察了反应时间、PS浓度和外加NH4+对硝基副产物生成的影响,同时对NH4+的转化途径以及硝基副产物的生成机制进行了探讨.本研究结果可为评估过硫酸盐高级氧化技术的可行性提供依据.
1 材料与方法
1.1 材料与试剂
所有试剂均为分析纯及以上级别.过硫酸钠(Na2S2O8)、硫酸铵、15N标记的硫酸铵(纯度99%)、3-硝基酚、4-硝基酚、4-羟基-3-硝基苯甲酸、2-羟基-5-硝基苯甲酸和2,4-二硝基酚购于阿拉丁(上海).色谱甲醇和甲酸购于TEDIA(Fairfield,美国).硫酸(H2SO4,纯度98.3%)、氢氧化钠、氯化钾、重铬酸钾、氯化钡和硫酸镁购自国药沪试(上海).C18固相萃取小柱(6cc/500mg, D110534)购自月旭科技(上海).所有实验用水均为超纯水(18.2MΩ×cm),由Stakpure OmniaTap水净化系统制备得到(Peculiar Instrument Technology Limited,英国).
供试土壤分别采集于江苏南京农业大学白马科研基地(119.185521°E,31.614205°N)和河北省邯郸市曲周县(114.988701°E,36.807845°N),采样深度为0~20cm.将土壤样品磨碎后过10目筛,风干老化后再研磨,过20目筛,分装保存.取10g土壤与一定量纯水混合(土水质量比为1:2.5),振荡5min,静置24h后测定pH值.土壤有机质(SOM)的含量通过重铬酸钾容量法(外加热法)测定[37].土壤阳离子交换能力(CEC)通过BaCl2-MgSO4交换法测定[37].土壤NO3--N和NH4+-N用2mol/L氯化钾溶液浸提后,采用FIAstar 5000流动分析仪(Foss Tecator,丹麦)测定[37].供试土壤的理化性质见表1.
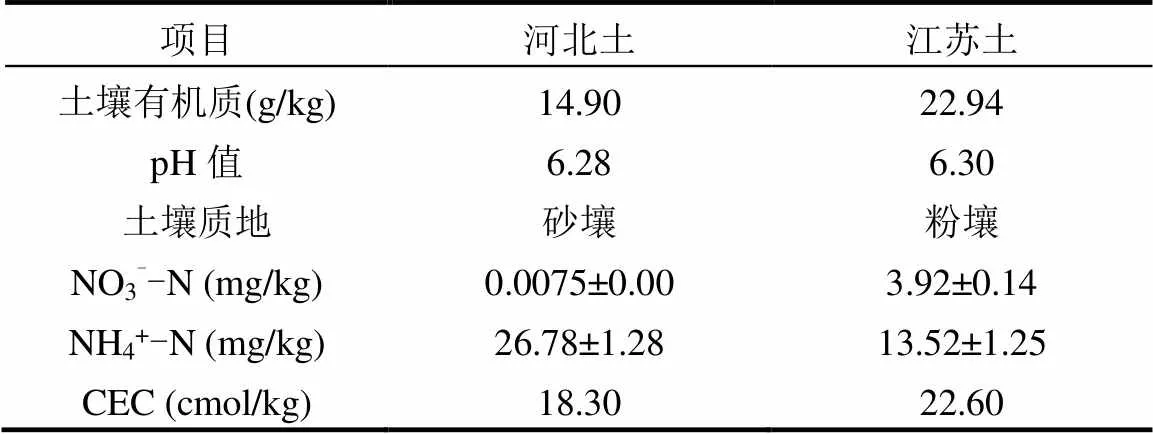
表1 供试土壤的理化性质
1.2 实验设计
反应在50mL离心管内进行,每组设置3个平行.每个样品含有10g土壤样本,然后加入15mL一定浓度的PS溶液,PS浓度设7.5, 15, 30, 45, 75mmol/kg 5个水平.在处理江苏土过程中,额外加入7.5mmol/kg NH4+,其中一组加入物质的量浓度1:1的14NH4+和15NH4+,另一组只加入14NH4+.反应前将摇床预热至60℃,随后将充分混匀的样品放入摇床内反应,转速设为250r/min.分别于0, 3, 6, 9, 12, 15, 18, 21, 24h取出样品,置于冰水浴冷却,终止反应.样品经过3000r/min离心10min,进行固液分离.上清液加入H2SO4酸化至pH<2,然后用C18固相萃取(SPE)小柱进行富集.在萃取前,SPE小柱依次用5mL甲醇和5mL 酸水(pH值 3)进行活化,随后上样.样品富集后,先后用2mL水和2mL 体积分数为3%的甲醇洗去杂质,加压吹干.最后将富集在柱内的样品用2mL甲醇洗脱,氮吹浓缩至1mL,过0.45μm滤膜(有机尼龙66,津隆),转移至液相小瓶中,4℃保存待分析.固相样品中继续加入10mL氢氧化钠溶液使pH值>13,超声30min萃取,3000r/min离心10min,取上清液.重复该过程2次,合并萃取液,加H2SO4酸化后,用SPE富集,步骤同上.
此外,为了研究外加NH4+对硝基副产物生成的影响,在10g河北土壤样本中加入15mL含有一定量PS和NH4+的混合溶液,使得PS浓度为30mmol/kg,外加NH4+浓度分别为0, 1.5, 3.0, 4.5, 6.0, 7.5mmol/ kg.其余反应条件同上,24h后冰水浴终止反应.样品处理过程同上.
1.3 硝基副产物的定性、定量分析
参考先前报道的方法对硝基副产物进行定性和定量分析[20-21].富集后的样品通过高效液相色谱-质谱联用仪(HPLC-MS/MS)进行分析.液相色谱型号为Agilent 1200,配有Agilent Zorbax Eclipse Plus C18反相柱(150mm × 2.1mm × 3.5 μm),流动相为甲醇与水(均加入0.1%体积甲酸酸化),流速0.2mL/min.前10min,甲醇体积分数从20%上升至30%;10~ 20min,甲醇体积分数从30%上升至70%;20~25min,甲醇体积分数从70%上升至100%并保持5min.MS检测器型号为Agilent 6410,配备电喷雾离子化接口(ESI)和三重四极杆质量分析器,设为负离子模式,其它主要参数为:毛细管电压-4.0kV;碎裂电压125V;氮气(³99.995%)作干燥气,流速10mL/min;温度350℃;喷雾器压力40psi.根据标样的二级质谱选择特征的母子离子对(表2),采用多反应监测模式(MRM),通过比较产物与标样的保留时间及峰面积,确定产物的结构和浓度.
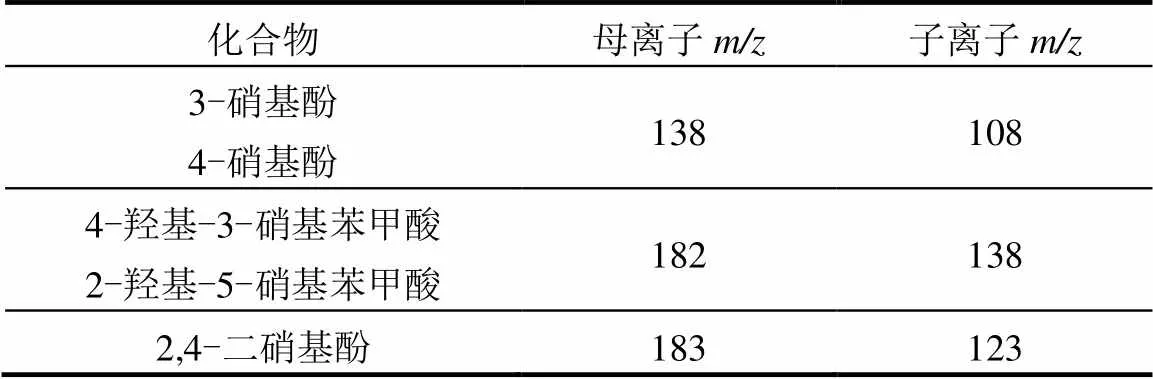
表2 硝基副产物HPLC-MS/MS分析的离子对选择和碰撞能量
1.4 硝基副产物的回收率
参考先前报道的方法对土样上清液中硝基副产物的回收率进行测定[21].在50mL离心管内加入10g未经过处理的江苏土及15mL水,充分混匀后置于摇床内,转速设为250r/min,温度为室温.24h后3000r/min离心10min,取上清液.在上清液中加入一定量3-硝基酚、4-硝基酚、4-羟基-3-硝基苯甲酸和2-羟基-5-硝基苯甲酸的混合标样,使得各个标样的浓度为5μmol/L.随后加入H2SO4酸化,用SPE富集,并用MS进行分析.SPE步骤和MS分析方法同上.各个物质的浓度利用外标法进行测定,将测定浓度除以加标值计算得到回收率.3-硝基酚、4-硝基酚、4-羟基-3-硝基苯甲酸和2-羟基-5-硝基苯甲酸的回收率分别为53.16%±1.41%、39.50%±0.61%、94.19%±2.11%和91.59%±3.05%.样品中硝基副产物的浓度通过回收率进行校正.
2 结果与讨论
2.1 硝基副产物的鉴定
河北土有机质含量为14.90g/kg,NH4+-N含量为26.78mg/kg(相当于2mmol/kg).经过热活化PS处理后,土样上清液的质谱如图1所示.根据对谱图的分析,并结合已有研究表明,质谱峰138、154、182和183可能对应于含氮副产物.根据二级质谱并结合分子量,推测138为硝基酚,进一步利用HPLC-MS/MS分离并与标样进行比对,确定为3-硝基酚和4-硝基酚,其中4-硝基酚占主导;182对应于一硝基羟基苯甲酸,同样有2种同分异构体,分别为2-羟基-5-硝基苯甲酸和4-羟基-3-硝基苯甲酸,其中4-羟基-3-硝基苯甲酸占主导;183对应于2,4-二硝基酚.此外,根据分子量,推测154为二羟基硝基苯,但由于缺乏标准物质,无法进行进一步的结构确认.以上实验结果均由土样上清液测定得到,土样固相中硝基副产物的生成低于检测限,故后续实验结果均为土样上清液.
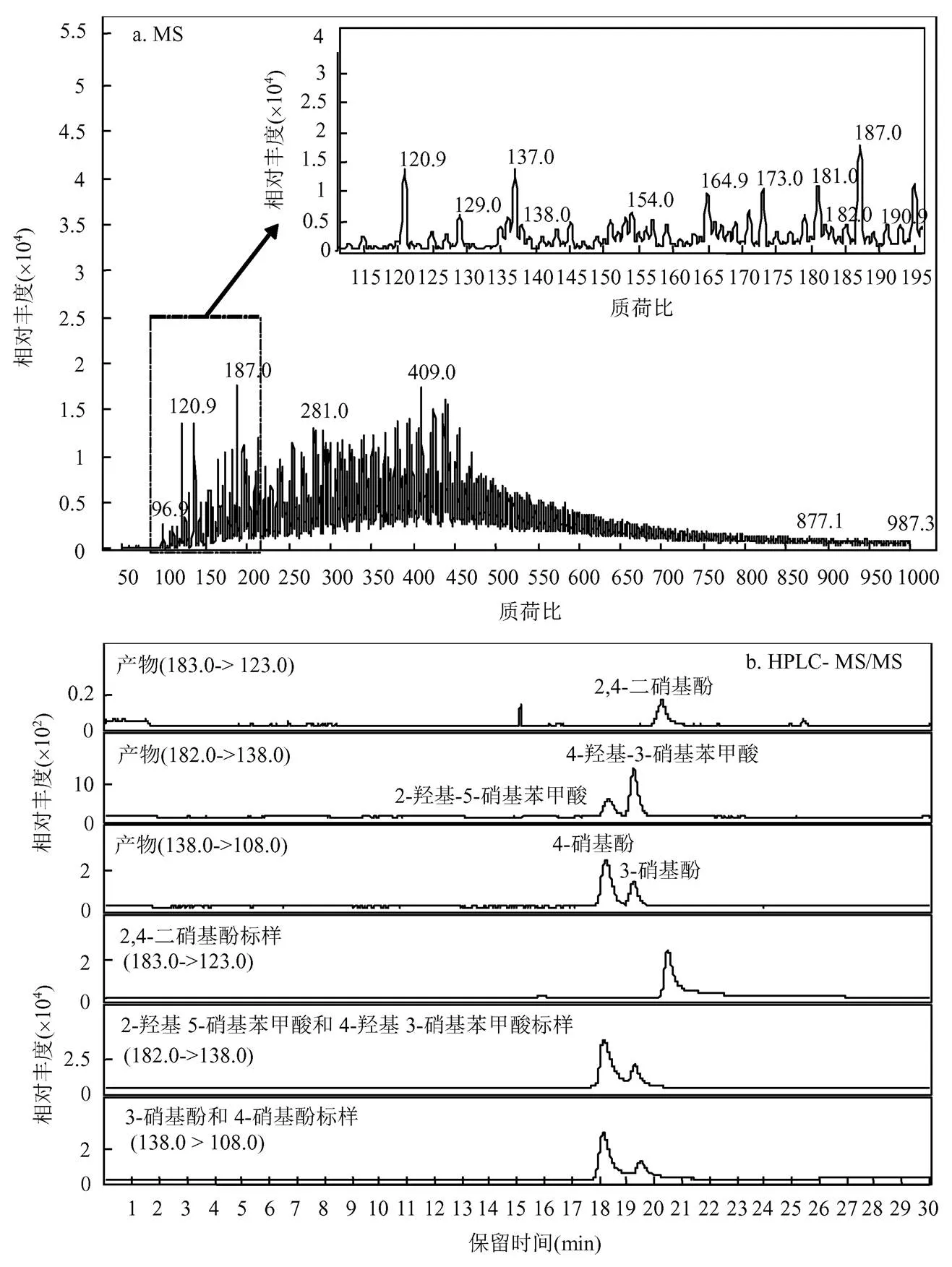
图1 热活化PS处理河北土土样上清液质谱和色谱分离图
反应条件:河北土10g,反应液15mL, PS 30mmol/kg, 60℃, 250r/min, 12h
江苏土有机质含量为22.94g/kg,NH4+-N含量为13.52mg/kg(相当于1mmol/kg).经过热活化PS处理后没有检测到明显的含氮副产物,这可能与土壤中NH4+含量较低有关.往土壤中加入7.5mmol/kg物质的量浓度1:1的14NH4+和15NH4+后,经过同样的处理,并通过MS图谱中的15N特征同位素指纹来判断是否生成了含氮副产物.由于反应含有物质的量浓度1:1的14NH4+和15NH4+,因此,NH4+转化为有机氮后的产物含有相应的同位素特征,即伴随有(+1)、(+2)…(+)的同位素峰(其中代表分子中含有来自NH4+的N原子数量),它们的相对峰度比值符合二项式(1+1)的展开.
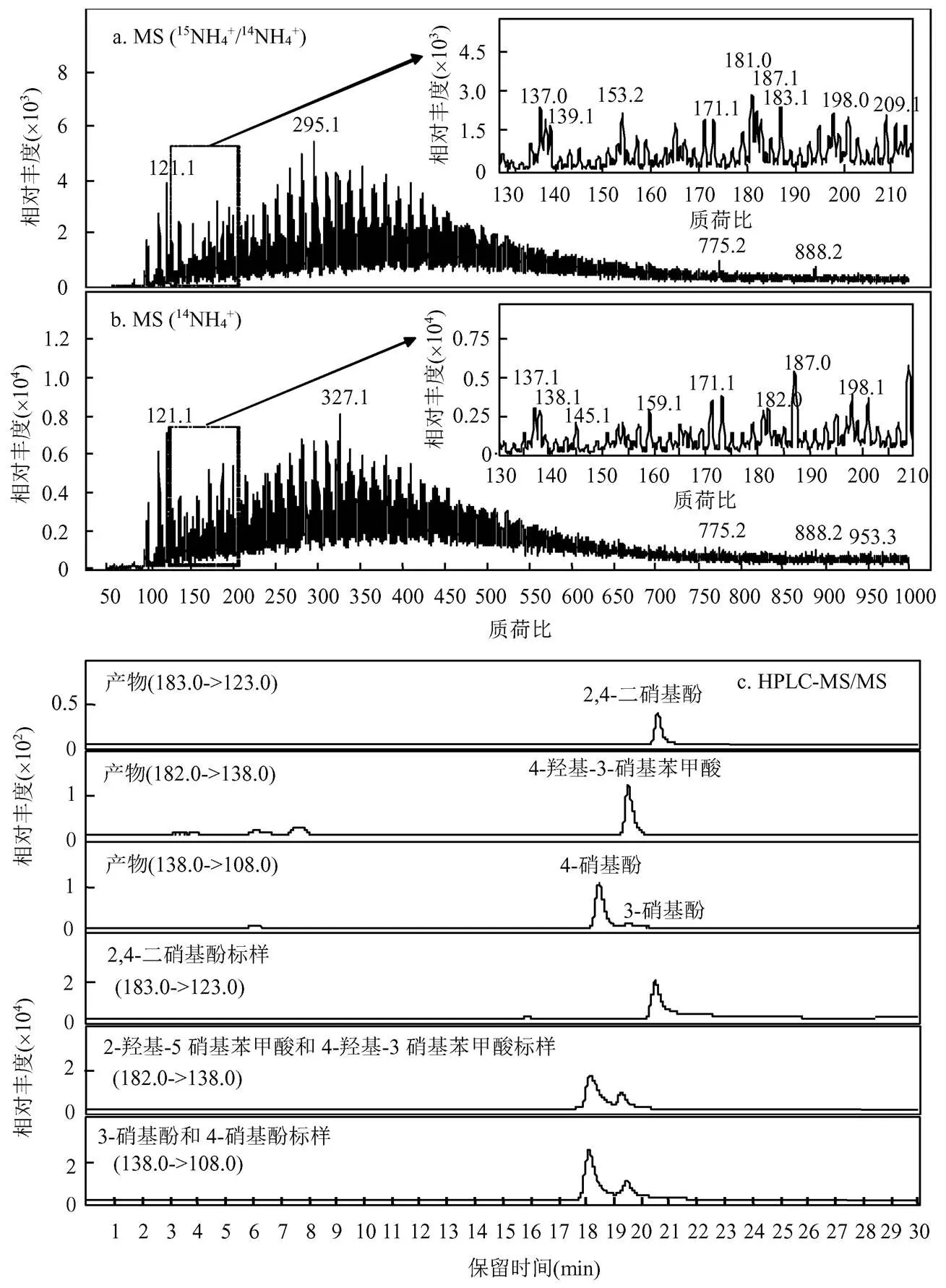
图2 热活化PS处理江苏土土样上清液质谱和色谱分离图
反应条件:江苏土10g,反应液15mL, PS 30mmol/kg, NH4+7.5mmol/kg, 60℃, 250r/min, 12h,其中a为加入物质的量浓度1:1的14NH4+和15NH4+, b为只加入14NH4+
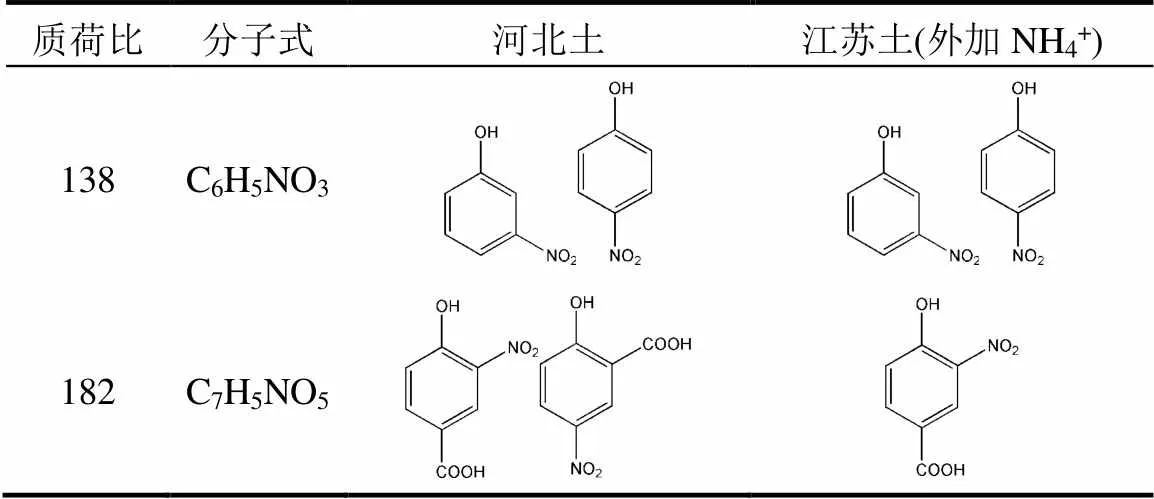
表3 热活化PS处理后,土壤中检测到的硝基副产物
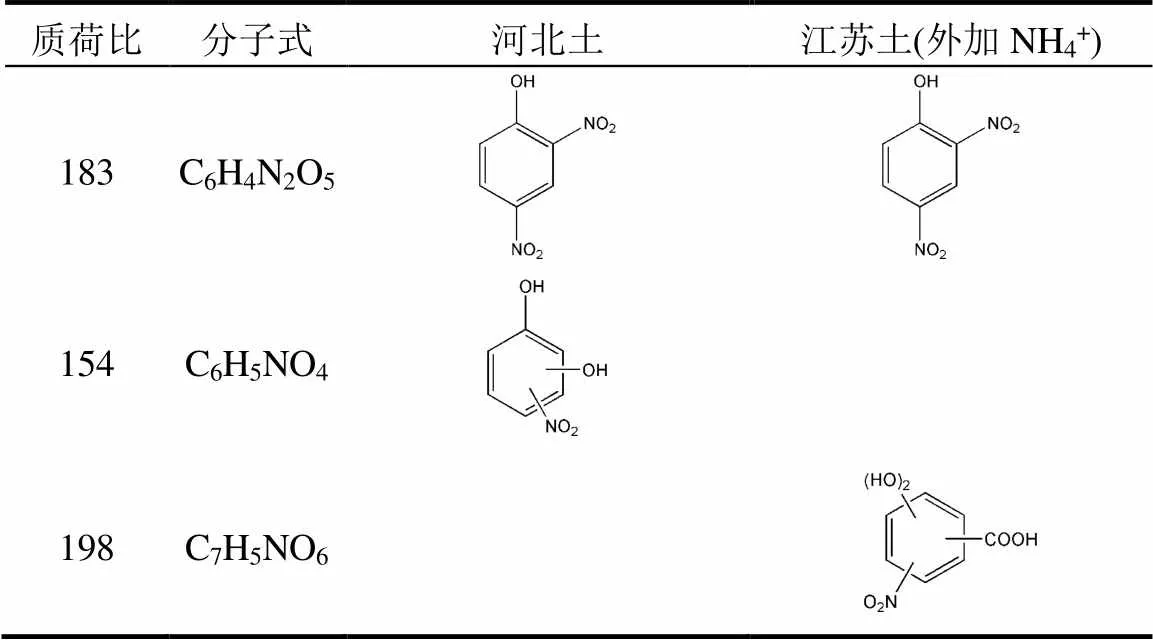
续表3
如图2a所示,反应后出现了138和139质谱峰,且峰度比接近1:1,而只加入14NH4+实验组中只有138质谱峰(图2b),说明该物质含有一个氮原子,根据分子量推测138为一硝基酚.同样地,利用HPLC-MS/MS分离并与标样进行比对,138确定为3-硝基酚和4-硝基酚,其中4-硝基酚占主导(图2c).利用上述方法,进一步确定182为4-羟基-3-硝基苯甲酸;183为2,4-二硝基酚.此外,根据15N同位素指纹结合分子量,推测198为二羟基硝基苯甲酸,但由于缺乏标准物质,无法进行进一步的结构确认.综上所述,热活化PS氧化土壤过程中,NH4+能够转化生成硝基副产物,它们的分子式与结构见表3.
2.2 河北土中硝基副产物的生成
对样品中的硝基副产物浓度进行定量分析,如图3所示,一硝基羟基苯甲酸的浓度高于一硝基酚.当PS浓度为30mmol/kg时,硝基副产物的生成量随着反应的进行先增加后降低,在反应12h达到最大值,其中一硝基酚和一硝基羟基苯甲酸浓度分别为0.0044和0.0072μmol/kg(图3a).2,4-二硝基酚生成量较低,故后续将不对其进行定量分析.此外,二羟基硝基苯(154)由于缺乏标准物质无法定量,但其峰面积也随着反应的进行先增加后降低.
如图3b所示,硝基副产物的生成量随着PS浓度的增加先增加后降低.在反应12h条件下,当PS浓度为30mmol/kg时,一硝基酚和一硝基羟基苯甲酸的生成量达到最大;当PS浓度为60mmol/kg时,二羟基硝基苯(154)的峰面积达到最大.研究表明,SO4×-通过电子转移能够将NOM大分子中的酚结构单元氧化成苯氧自由基,苯氧自由基进一步与NO2×结合生成硝基副产物[20-23].因此,增大PS浓度有利于SO4×-的生成,一方面促进NH4+转化生成NO2×,另一方面氧化土壤有机质生成苯氧自由基,最终促进硝基副产物的生成.当PS浓度继续增大,硝基副产物可能进一步被SO4×-氧化.
当PS浓度为30mmol/kg,往土壤中加入不同浓度的NH4+后,延长反应时间至24h.如图3c所示,硝基副产物的生成量随着NH4+浓度的增加先增加后降低.当NH4+加入量为1.5mmol/kg时,硝基副产物生成量达到最大,其中一硝基酚和一硝基羟基苯甲酸的浓度分别为0.0056和0.0125μmol/kg.结果表明,提高NH4+浓度有利于NO2×的生成,并进一步转化生成硝基副产物.当NH4+浓度继续增加时,NH4+可能与土壤有机质竞争SO4×-,使得苯氧自由基中间体的生成受到抑制,从而导致硝基副产物生成量下降.

反应条件: a.河北土10g,反应液15mL, PS 30mmol/kg, 60℃, 250r/min; b.河北土10g,反应液15mL, 60℃, 250r/min, 12h; c.河北土10g,反应液15mL, PS 30mmol/kg, 60℃, 250r/min, 24h. 柱状代表可定量的硝基副产物,其浓度对应左纵坐标轴;折线代表不可定量的硝基副产物,其峰面积对应右纵坐标轴
2.3 江苏土中硝基副产物的生成
与河北土相似,江苏土中一硝基羟基苯甲酸的浓度高于一硝基酚.如图4a所示,在外加NH4+浓度为7.5mmol/kg、PS浓度为30mmol/kg条件下,硝基副产物的生成量随着反应的进行先增加后降低,在反应12h达到最大值,其中一硝基酚和一硝基羟基苯甲酸浓度分别为0.0069和0.0186 μmol/kg.2,4-二硝基酚生成量较低,故后续将不对其进行定量分析.此外,二羟基硝基苯甲酸(198)由于缺乏标准物质无法定量,但其峰面积也随着反应的进行先增加后降低.
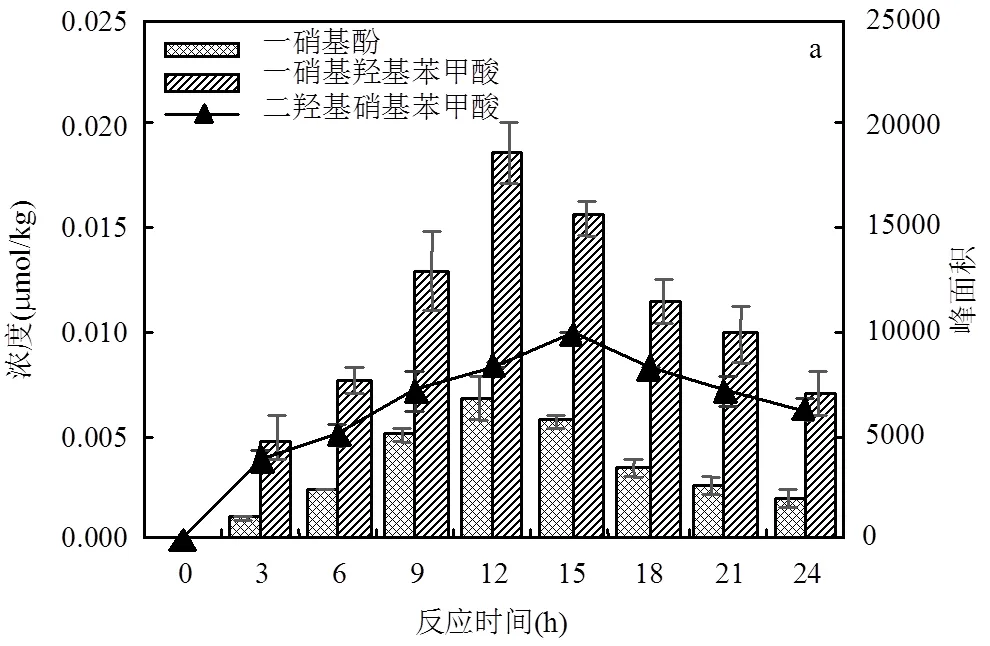
反应条件: a.江苏土10g,反应液15mL, PS 30mmol/kg, NH4+7.5mmol/kg,60℃, 250r/min; b.江苏土10g,反应液15mL, NH4+7.5mmol/kg, 60℃, 250r/min, 12h.柱状代表可定量的硝基副产物,其浓度对应左纵坐标轴;折线代表不可定量的硝基副产物,其峰面积对应右纵坐标轴
如图4b所示,硝基副产物的生成量随着PS浓度的增加先增加后降低.在反应12h、外加7.5mmol/kg NH4+条件下,当PS浓度为30mmol/kg时,一硝基酚、一硝基羟基苯甲酸和二羟基硝基苯甲酸的生成量达到最大.结果表明,增大PS浓度有利于SO4×-的生成,既可以促进NH4+转化生成NO2×,又能够氧化土壤有机质生成苯氧自由基,最终促进硝基副产物的生成.当PS浓度继续增大,硝基副产物可能进一步被SO4×-氧化.以上实验结果与河北土中硝基副产物的生成一致.
2.4 NH4+的转化路径
以上实验表明,NH4+在SO4×-氧化过程中的确发生了转化.参考NH4+在×OH反应系统中的转化路径,推测NH4+首先与SO4×-反应生成×NH2,两者反应速率为3×105mol/(L×s)[25-26].且×OH常常存在于SO4×-氧化过程中,在中性或酸性条件下,×OH浓度较低[38],但也能够氧化NH4+生成×NH2[25].×NH2在有氧条件下能够被迅速氧化为NH2O2×,经重排分解后生成NO×,继而被×OH氧化生成NO2-[25-26,33].NO2-能够被SO4×-或×OH氧化生成NO2×[34,39].此外,NO×也能够被氧气直接氧化生成NO2×[33].NO2×自身耦合形成N2O4,后者能进一步水解生成NO3-[34].值得注意的是,一旦NH4+的转化过程中能够生成NO2×,若存在天然有机质,NO2×能够和天然有机质反应生成硝基副产物.如图5所示,对于额外加入NH4+的江苏土,反应前NH4+-N浓度为8.47mmol/kg,NO3--N仅为0.24mmol/kg,总无机氮浓度为8.71mmol/kg.当PS浓度为30mmol/kg,反应12 与24h后,NH4+-N分别减少了1.31和2.27mmol/kg,而NO3--N分别增加了0.64和1.12mmol/kg,没有检测到NO2-,土壤中总无机氮逐渐降低至8.04和7.56mmol/kg.而此时,江苏土中检测到的硝基副产物中氮含量分别为0.0255和0.009 μmol/kg.由此可见,大部分NH4+-N在热活化PS处理土壤过程中转化为NO3--N,仅有少量转化成有机氮.此外,从N质量守恒来看,随着反应的进行,样品中的总氮在不断减少.有研究表明,×NH2自身耦合生成肼(N2H4)或与×OH结合生成羟胺(NH2OH),两者均能在PS的作用下转化为N2逸出[40-42].另外,本样品中还有部分硝基副产物由于缺乏标准物质而无法进行定量分析.综上所述,活化过硫酸盐氧化过程中NH4+转化路径见图6.
河北土中NH4+-N浓度为1.91mmol/kg,而NO3--N浓度几乎为0.经过同样条件处理,土壤中的总无机氮逐渐降低至1.40和1.16mmol/kg,其中NH4+-N在12h时转化了1.17mmol/kg,24h时几乎完全转化,而NO3--N浓度分别增加至0.66与1.08mmol/kg,同样没有检测到NO2-.此外,反应12与24h后,河北土中硝基副产物的氮含量分别为0.0116和0.0051 μmol/kg.结果表明,大部分NH4+-N在热活化PS处理土壤过程中转化为NO3--N,少量能够转化成有机氮,仍有一部分N没有被检测到.上述实验结果与江苏土实验结果一致.
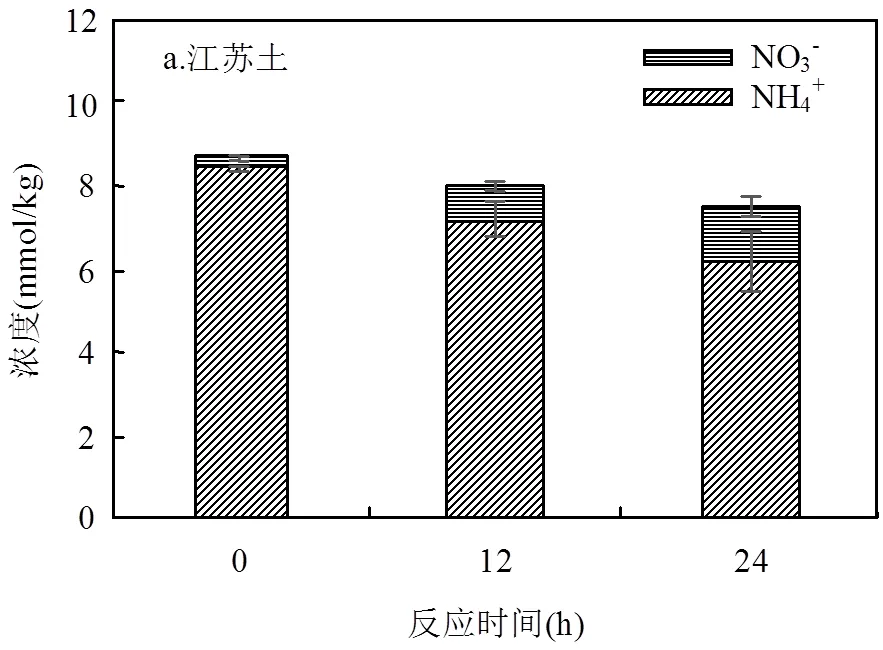
反应条件: a.江苏土10g,反应液15mL, PS 30mmol/kg, NH4+7.5mmol/kg, 60℃, 250r/min; b.河北土10g,反应液15mL, PS 30mmol/kg, 60℃, 250r/min
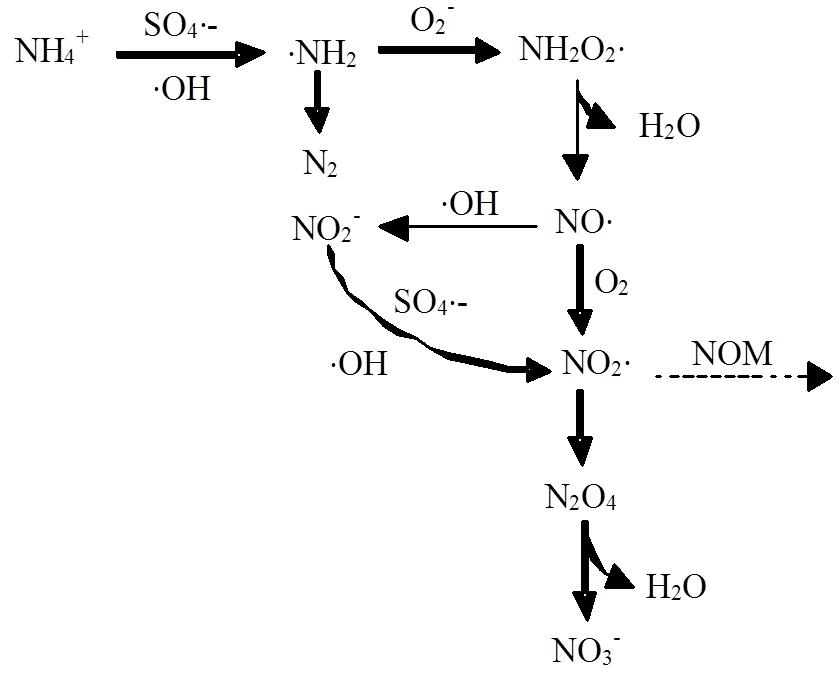
图6 活化过硫酸盐氧化过程中NH4+的转化路径
3 结论
3.1 经热活化PS处理后,河北土和江苏土中均检测出了一硝基酚、一硝基羟基苯甲酸、二硝基酚等副产物.这是由于土壤中的铵氮被SO4×-氧化生成了×NH2,并通过自由基链式反应生成NO2×.NO2×能够与土壤有机质反应,生成硝基副产物.
3.2 两种土壤样本中硝基副产物的生成量在反应12h、PS浓度为30mmol/kg时达到最大,且这些硝基副产物大多分布于土样上清液中,这意味着它们具有较强的迁移性,可随着地表径流转移或流入地下,造成更大范围的污染.
[1] Tsitonaki A, Petri B, Crimi M, et al.chemical oxidation of contaminated soil and groundwater using persulfate: A review [J]. Critical Reviews in Environmental Science and Technology, 2010, 40(1):55-91.
[2] Long A H, Lei Y, Zhang H.chemical oxidation of organic contaminated soil and groundwater using activated persulfate process [J]. Progress in Chemistry, 2014,26(5):898-908.
[3] Zhang B, Zhang Y, Teng Y, et al. Sulfate radical and its application in decontamination technologies [J]. Critical Reviews in Environmental Science and Technology, 2015,45(16):1756-1800.
[4] Matzek L W, Carter K E. Activated persulfate for organic chemical degradation: A review [J]. Chemosphere, 2016,151:178-188.
[5] Zhou Y, Xiang Y, He Y, et al. Applications and factors influencing of the persulfate-based advanced oxidation processes for the remediation of groundwater and soil contaminated with organic compounds [J]. Journal of Hazardous Materials, 2018,359:396-407.
[6] Neta P, Huie R, Ross A. Rate constants for reactions of inorganic radicals in aqueous solution [J]. Journal of Physical and Chemical Reference Data, 1988,17(3):1027-1284.
[7] Liang C, Lee I L, Hsu I Y, et al. Persulfate oxidation of trichloroethylene with and without iron activation in porous media [J]. Chemosphere, 2008,70(3):426-435.
[8] Usman M, Faure P, Ruby C, et al. Application of magnetite-activated persulfate oxidation for the degradation of PAHs in contaminated soils [J]. Chemosphere, 2012,87(3):234-240.
[9] Qian Y, Guo X, Zhang Y, et al. Perfluorooctanoic acid degradation using UV-persulfate process: Modeling of the degradation and chlorate formation [J]. Environmental Science & Technology, 2016,50 (2):772-781.
[10] Bruton T A, Sedlak D L. Treatment of perfluoroalkyl acids by heat-activated persulfate under conditions representative ofchemical oxidation [J]. Chemosphere, 2018,206:457-464.
[11] Waldermer R, Tratnyek P, Johnson R, et al. Oxidation of chlorinated ethenes by heat activated persulfate: Kinetics and products [J]. Environmental Science & Technology, 2007,41(3):1010-1015.
[12] 刘小宁.利用热活化过硫酸盐修复氯苯污染地下水的研究[D]. 上海:华东理工大学, 2013.
Liu X N. Remediation of chlorobenzene-contaminated groundwater by thermally activated persulfate [D]. Shanghai:East China University of Science and Technology, 2013.
[13] Liu Y, Wang S, Wu Y, et al. Degradation of ibuprofen by thermally activated persulfate in soil systems [J]. Chemical Engineering Journal, 2019,356:799-810.
[14] 吴承梓,张 巍,万彦涛,等.盐酸羟胺/铁基MOFs/过硫酸盐体系降解磺胺嘧啶 [J]. 中国环境科学, 2021,41(6):2685-2697.
Wu C Z, Zhang W, Wan Y T, et al. Degradation of sulfadiazine by hydroxylamine hydrochloride/Fe-MOFs/persulfate system [J].China Environmental Science, 2021,41(6):2685-2697.
[15] Lu J, Wu J, Ji Y, et al. Transformation of bromide in thermo activated persulfate oxidation processes [J].Water Research, 2015,78:1-8.
[16] Fang J, Shang C. Bromate formation from bromide oxidation by the UV/persulfate process [J]. Environmental Science & Technology, 2012,46(16):8976-8983.
[17] Wang Y, Roux J L, Zhang T, et al. Formation of brominated disinfection byproducts from natural organic matter isolates and model compounds in a sulfate radical-based oxidation process [J]. Environmental Science & Technology, 2014,48(24):14534-14542.
[18] Wang L, Kong D, Ji Y, et al. Transformation of iodide and formation of iodinated by-products in heat activated persulfate oxidation process [J].Chemosphere, 2017,181:400-408.
[19] Ji Y, Wang L, Jiang M, et al. The role of nitrite in sulfate radical-based degradation of phenolic compounds: An unexpected nitration process relevant to groundwater remediation bychemical oxidation (ISCO) [J]. Water Research, 2017,123:249-257.
[20] Yang P, Ji Y, Lu J, et al. Formation of nitrophenolic byproducts during heat-activated peroxydisulfate oxidation in the presence of natural organic matter and nitrite [J]. Environmental Science & Technology, 2019,53(8):4255-4264.
[21] Yang P, Qian L, Cheng Y, et al. Formation of nitrophenolic byproducts in soils subjected to sulfate radical oxidation [J]. Chemical Engineering Journal, 2021,403:126316.
[22] Barzaghi P, Herrmann H. A mechanistic study of the oxidation of phenol by OH/NO2/NO3in aqueous solution [J]. Physical Chemistry Chemical Physics, 2002,4(15):3669-3675.
[23] Bedini A, Maurino V, Minero C, et al. Theoretical and experimental evidence of the photonitration pathway of phenol and 4-chlorophenol: A mechanistic study of environmental significance [J]. Photochemical & Photobiological Sciences, 2012,11(2):418-424.
[24] Zhang Q, Ren F, Li F, et al. Ammonia nitrogen sources and pollution along soil profiles in anleaching rare earth ore [J]. Environmental Pollution, 2020,267:115449.
[25] Laszlo B, Alfassi Z B, Neta P, et al. Kinetics and mechanism of the reaction of •NH2with O2in aqueous solutions [J]. Journal of Physical Chemistry A, 1998,102:8498-8504.
[26] Pagsberg P B. Investigation of the NH2radical produced by pulse radiolysis of ammonia in aqueous solution [R]. Riso National Laboratory Report, Roskilde, Denmark, 1972,256:209-221.
[27] Clarke K, Edge R, Johnson V L, et al. Direct observation of •NH2reactions with oxygen, amino acids, and melanins [J]. Journal of Physical Chemistry A, 2008,112(6):1234-1237.
[28] Huang L, Li L, Dong W, et al. Removal of ammonia by OH radical in aqueous phase [J]. Environmental Science & Technology, 2008,42: 8070-8075.
[29] Dwibedy P, Kishore K, Dey G R, et al. Nitrite formation in the radiolysis of aerated aqueous solutions of ammonia [J]. Radiation Physics and Chemistry, 1996,48:743-747.
[30] Wang J, Song M, Chen B, et al. Effects of pH and H2O2on ammonia, nitrite, and nitrate transformations during UV254nm irradiation: Implications to nitrogen removal and analysis [J]. Chemosphere, 2017,184:1003-1011.
[31] Zhang X, Ren P, Li W, et al. Synergistic removal of ammonium by monochloramine photolysis [J]. Water Research, 2019,152:226-233.
[32] Stanbury D M. Reduction potentials involving inorganic free radicals in aqueous solution [J]. Advances in Inorganic Chemistry, 1989,33:69-138.
[33] Wu Z, Chen C, Zhu B Z, et al. Reactive nitrogen species are also involved in the transformation of micropollutants by the UV/ monochloramine Process [J]. Environmental Science & Technology, 2019,53(19):11142-11152.
[34] John M, James R B. Photochemistry of nitrite and nitrate in aqueous solution: A review [J]. Journal of Photochemistry and Photobiology A: Chemistry, 1999,128:1-13.
[35] De Laat J, Boudiaf N, Dossier-Berne F. Effect of dissolved oxygen on the photodecomposition of monochloramine and dichloramine in aqueous solution by UV irradiation at 253.7nm [J]. Water Research, 2010,44(10):3261-3269.
[36] Dey G R. Nitrogen compounds' formation in aqueous solutions under high ionizing radiation: An overview [J]. Radiation Physics and Chemistry, 2011,80(3):394-402.
[37] 鲍士旦.土壤农化分析[M]. 3版.北京:中国农业出版社, 2013.
Bao S D. Soil agrochemical analysis [M]. Third Edition. Beijing: China Agriculture Press, 2013.
[38] Liang C, Su H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate [J]. Industrial & Engineering Chemistry Research, 2009,48:5558-5562.
[39] Neta P, Madhavan V, Zemel H, et al. Rate constants and mechanism of reaction of sulfate radical anion with aromatic compounds [J]. Journal of the American Chemical Society, 1977,99(1):163-164.
[40] Gonzalez M C, Braun A M. Vacuum-UV photolysis of aqueous solutions of nitrate: Effect of organic matter I. Phenol [J]. Journal of Photochemistry and Photobiology A: Chemistry, 1996,93:7-19.
[41] Feng Y, Wu D, Zhou Y, et al. A metal-free method of generating sulfate radicals through direct interaction of hydroxylamine and peroxymonosulfate: Mechanisms, kinetics, and implications [J]. Chemical Engineering Journal, 2017,330:906-913.
[42] Ball, A, Chako J O, Edwards G L. Mechanisms of oxidation of nitrogen nucleophiles by peroxodisulfate ion: Nitrite ion and ammonia [J]. Inorganica Chimica Acta, 1985,99:49-58.
Transformation of soil ammonium nitrogen in the process of thermally activated persulfate oxidation.
YANG Pei-zeng, YUE Hong-shen, JI Yue-fei, LU Jun-he*
(College of Resource and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095, China)., 2022,42(1):267~275
In order to explore the transformation and fate of soil NH4+in the thermally activated PS oxidation process, this study used soil samples collected from Jiangsu and Hebei provinces with different soil organic matter content and NH4+concentration to conduct experiments, and systematically investigated effects of persulfate (PS) concentration, the addition of NH4+, and reaction time on the formation of nitro by-products. Results show that soil NH4+could be transformed to nitrated byproducts, including 3-nitrophenol, 4-nitrophenol, 2-hydroxy-5-nitrobenzoic acid, 4-hydroxy-3-nitrobenzoic acid, 2,4-dinitrophenol, etc. The formation of nitro by-products increased first and then decreased with reaction time. An increased in PS dose would promote the formation of nitro by-products, and the yields of mono-nitrophenols and hydroxy-mono-nitrobenzoic acids reached the maximum after 12h reaction at 30mmol/kg PS dose. However, nitrated byproducts were degraded at higher PS dose. Note that sulfate radicals (SO4•-) played a key role in the nitration process by oxidizing NH4+to form aminyl radicals (•NH2), and then underwent a series of free radical chain reactions to form nitrogen dioxide radicals (NO2•). Besides, phenol moieties in soil organic matter served as the main reactive sites for SO4•-attack, leading to the formation of phenoxy radicals, which further combined with NO2• to form nitro by-products. NOM is everywhere and NH4+is ubiquitous in the environment. Thus, the formation of nitro by-products will be widespread when PS is applied for contaminatedsoil and groundwaterremediation, which should be taken into consideration when evaluating the feasibility of this technology. This study reveals that the presence of soil NH4+in activated PS oxidation processes could induce the nitration of NOM and the formation of nitrophenolic by-products.
ammonium;persulfate;soil organic matter;sulfate radical;nitrogen dioxide radical;nitrated byproducts
X703.5
A
1000-6923(2022)01-0267-09
杨培增(1994-),女,浙江嘉兴人,南京农业大学博士研究生,主要研究方向为水处理高级氧化.发表论文8篇.
2021-06-04
国家自然科学基金资助项目(22076079,22076080);江苏省研究生科研创新计划(030-Z562015603);国家大学生实践创新训练计划项目(201910307043Z)
* 责任作者, 教授, jhlu@njau.edu.cn
